

A proteome-wide genetic investigation identifies several SARS-CoV-2-exploited host targets of clinical relevance
- PMID: 34402426
- PMCID: PMC8457835
- DOI: 10.7554/eLife.69719
A proteome-wide genetic investigation identifies several SARS-CoV-2-exploited host targets of clinical relevance
Abstract
Background: The virus SARS-CoV-2 can exploit biological vulnerabilities (e.g. host proteins) in susceptible hosts that predispose to the development of severe COVID-19.
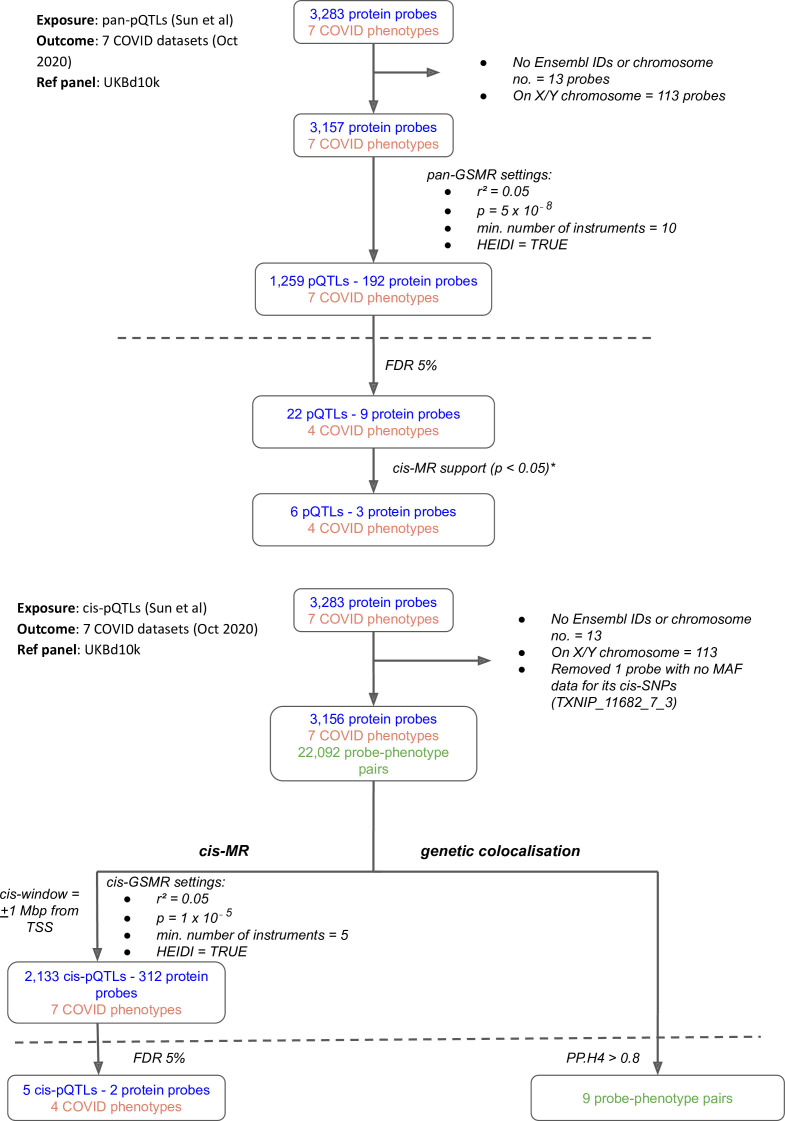
Methods: To identify host proteins that may contribute to the risk of severe COVID-19, we undertook proteome-wide genetic colocalisation tests, and polygenic (pan) and cis-Mendelian randomisation analyses leveraging publicly available protein and COVID-19 datasets.
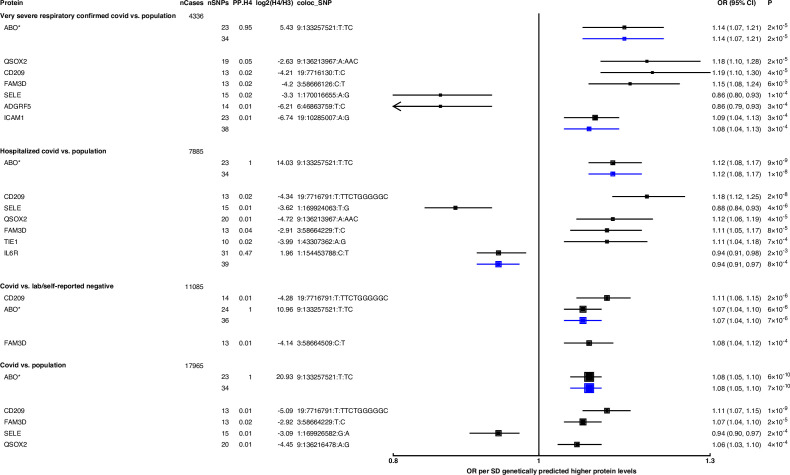
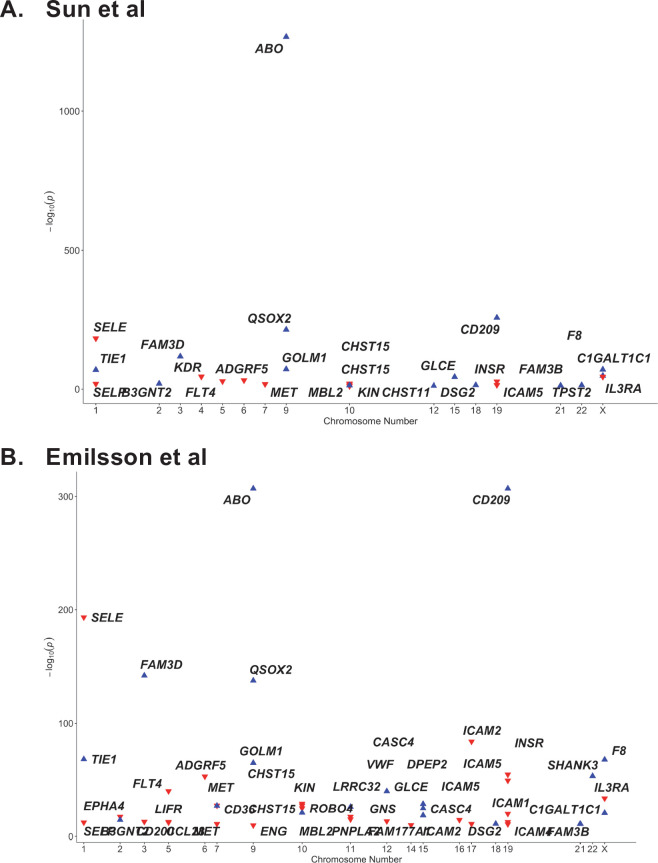
Results: Our analytic approach identified several known targets (e.g. ABO, OAS1), but also nominated new proteins such as soluble Fas (colocalisation probability >0.9, p=1 × 10-4), implicating Fas-mediated apoptosis as a potential target for COVID-19 risk. The polygenic (pan) and cis-Mendelian randomisation analyses showed consistent associations of genetically predicted ABO protein with several COVID-19 phenotypes. The ABO signal is highly pleiotropic, and a look-up of proteins associated with the ABO signal revealed that the strongest association was with soluble CD209. We demonstrated experimentally that CD209 directly interacts with the spike protein of SARS-CoV-2, suggesting a mechanism that could explain the ABO association with COVID-19.
Conclusions: Our work provides a prioritised list of host targets potentially exploited by SARS-CoV-2 and is a precursor for further research on CD209 and FAS as therapeutically tractable targets for COVID-19.
Funding: MAK, JSc, JH, AB, DO, MC, EMM, MG, ID were funded by Open Targets. J.Z. and T.R.G were funded by the UK Medical Research Council Integrative Epidemiology Unit (MC_UU_00011/4). JSh and GJW were funded by the Wellcome Trust Grant 206194. This research was funded in part by the Wellcome Trust [Grant 206194]. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Keywords: COVID-19; apoptosis; epidemiology; genetic colocalization; genetics; genomics; global health; human; mendelian randomization; proteins.
Plain language summary
Individuals who become infected with the virus that causes COVID-19 can experience a wide variety of symptoms. These can range from no symptoms or minor symptoms to severe illness and death. Key demographic factors, such as age, gender and race, are known to affect how susceptible an individual is to infection. However, molecular factors, such as unique gene mutations and gene expression levels can also have a major impact on patient responses by affecting the levels of proteins in the body. Proteins that are too abundant or too scarce may mean the difference between dying from or surviving COVID-19. Identifying the molecular factors in a host that affect how viruses can infect individuals, evade immune defences or trigger severe illness, could provide new ways to treat patients with COVID-19. Such factors are likely to remain constant, even when the virus mutates into new strains. Hence, insights would likely apply across all virus strains, including current strains, such as alpha and delta, and any new strains that may emerge in the future. Using such a ‘natural experiment’ approach, Karim et al. compared the genetic profiles of over 30,000 COVID-19 patients and a million healthy individuals. Nine proteins were found to have an impact on COVID-19 infection and disease severity. Four proteins were ranked as top priorities for potential treatment targets. One protein, called CD209 (also known as DC-SIGN), is involved in how the virus enters the host cells, and had one of the strongest associations with COVID-19. Two proteins, called IL-6R and FAS, were involved in the immune response and could be responsible for the immune over-activation often seen in severe COVID-19. Finally, one protein, called OAS1, formed part of the body’s innate antiviral defence system and appeared to reduce susceptibility to COVID-19. Knowing more about the proteins that influence the severity of COVID-19 opens up new ways to predict, protect and treat patients who may have severe or fatal reactions to infection. Indeed, one of the identified proteins (IL-6R) had already been targeted in recent clinical trials with some encouraging results. Considering CD209 as a potential receptor for the virus could provide another avenue for therapeutics, similar to previously successful approaches to block the virus’ known interaction with a receptor protein. Ultimately, this research could supply an entirely new set of treatment options to help combat the COVID-19 pandemic.
© 2021, Anisul et al.
Conflict of interest statement
MA, JS, JH, AB, JZ, DO, MC, VE, VG, EM, GW, MG Open Targets is a pre-competitive partnership currently involving the Wellcome Sanger Institute, EMBL-EBI, BMS, GSK, and Sanofi. Research is funded by financial and in-kind contributions from each of the partners. JS none, ES Open Targets is a pre-competitive partnership currently involving the Wellcome Sanger Institute, EMBL-EBI, BMS, GSK, and Sanofi. Research is funded by financial and in-kind contributions from each of the partners. ES is also a full-time employee of Bristol-Myers Squibb. MH Dr Holmes has consulted for Boehringer Ingelheim, and in adherence to the University of Oxford’s Clinical Trial Service Unit & Epidemiological Studies Unit (CSTU) staff policy, did not accept personal honoraria or other payments from pharmaceutical companies. JM JM is a full-time employee of Bristol-Myers Squibb and retains stock or stock options in Bristol-Myers Squibb. The author has no other competing interests to declare. TG TG received grants from Biogen and GlaxoSmithKline. The author has no other competing interests to declare. ID Open Targets is a pre-competitive partnership currently involving the Wellcome Sanger Institute, EMBL-EBI, BMS, GSK, and Sanofi. Research is funded by financial and in-kind contributions from each of the partners. ID also received travel costs within the last 36 months from Takeda for speaking at their Reverse Translation Symposium. The author has no other competing interests to declare.
Figures



Similar articles
-
Emerging Variants of SARS-CoV-2 and Novel Therapeutics Against Coronavirus (COVID-19).2023 May 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2023 May 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 34033342 Free Books & Documents.
-
Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants.Elife. 2020 Oct 28;9:e61312. doi: 10.7554/eLife.61312. Elife. 2020. PMID: 33112236 Free PMC article.
-
Comprehensive characterization of the antibody responses to SARS-CoV-2 Spike protein finds additional vaccine-induced epitopes beyond those for mild infection.Elife. 2022 Jan 24;11:e73490. doi: 10.7554/eLife.73490. Elife. 2022. PMID: 35072628 Free PMC article.
-
Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.Cochrane Database Syst Rev. 2022 Feb 1;2(2022):CD014217. doi: 10.1002/14651858.CD014217. Cochrane Database Syst Rev. 2022. PMID: 36321557 Free PMC article.
-
Variants in ACE2; potential influences on virus infection and COVID-19 severity.Infect Genet Evol. 2021 Jun;90:104773. doi: 10.1016/j.meegid.2021.104773. Epub 2021 Feb 17. Infect Genet Evol. 2021. PMID: 33607284 Free PMC article. Review.
Cited by
-
Interferon Control of Human Coronavirus Infection and Viral Evasion: Mechanistic Insights and Implications for Antiviral Drug and Vaccine Development.J Mol Biol. 2022 Mar 30;434(6):167438. doi: 10.1016/j.jmb.2021.167438. Epub 2022 Jan 3. J Mol Biol. 2022. PMID: 34990653 Free PMC article. Review.
-
Autoantibodies in hospitalised patients with COVID-19.Clin Transl Immunology. 2024 Dec 26;13(12):e70019. doi: 10.1002/cti2.70019. eCollection 2024. Clin Transl Immunology. 2024. PMID: 39734590 Free PMC article.
-
Plasma Proteome-Wide Mendelian Randomization Analysis Reveals Biomarkers and Therapeutic Targets for Different Stages of COVID-19.Transbound Emerg Dis. 2024 Feb 5;2024:5566180. doi: 10.1155/2024/5566180. eCollection 2024. Transbound Emerg Dis. 2024. PMID: 40303168 Free PMC article.
-
LRRC15 mediates an accessory interaction with the SARS-CoV-2 spike protein.PLoS Biol. 2023 Feb 3;21(2):e3001959. doi: 10.1371/journal.pbio.3001959. eCollection 2023 Feb. PLoS Biol. 2023. PMID: 36735681 Free PMC article.
-
Proteomic and metabolomic analysis of serum in women infected with COVID-19 during late pregnancy.Front Immunol. 2025 Jun 11;16:1589239. doi: 10.3389/fimmu.2025.1589239. eCollection 2025. Front Immunol. 2025. PMID: 40568583 Free PMC article.
References
-
- Amraie R. Cd209l/l-Sign and Cd209/Dc-Sign Act as Receptors for Sars-Cov-2 and Are Differentially Expressed in Lung and Kidney Epithelial and Endothelial Cells. bioRxiv. 2020 doi: 10.1101/2020.06.22.165803. - DOI
-
- Anisul M. Covid_paper. swh:1:rev:4ab9f9b17ffde57f7831ea555394290ba240a2b9Software Heritage. 2021 https://github.com/mohdkarim/covid_paper
-
- Anthony C G, Paul R M. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19 Preliminary Report. medRxiv. 2021 doi: 10.1101/2021.01.07.21249390. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
