

Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer
- PMID: 34404686
- PMCID: PMC8495771
- DOI: 10.1158/2159-8290.CD-21-0702
Parallel Genomic Alterations of Antigen and Payload Targets Mediate Polyclonal Acquired Clinical Resistance to Sacituzumab Govitecan in Triple-Negative Breast Cancer
Abstract
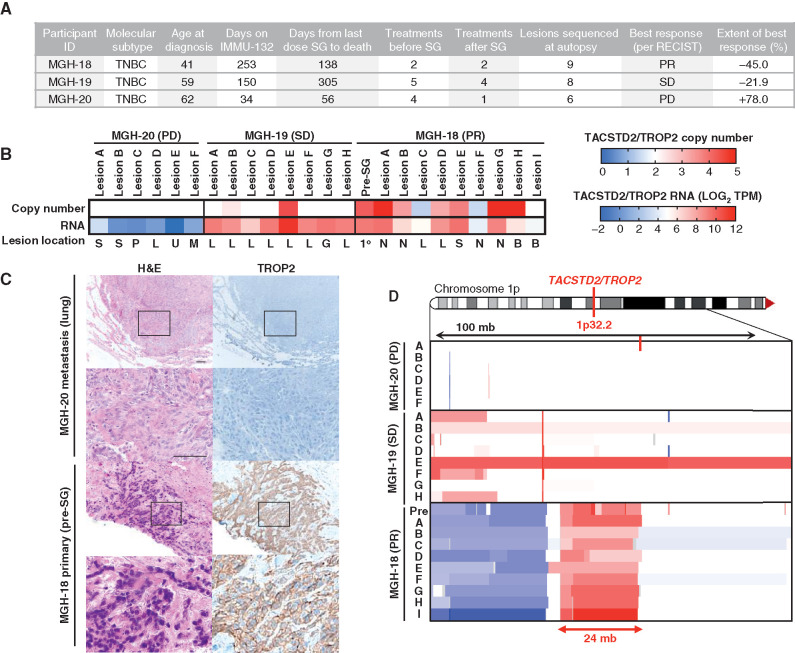
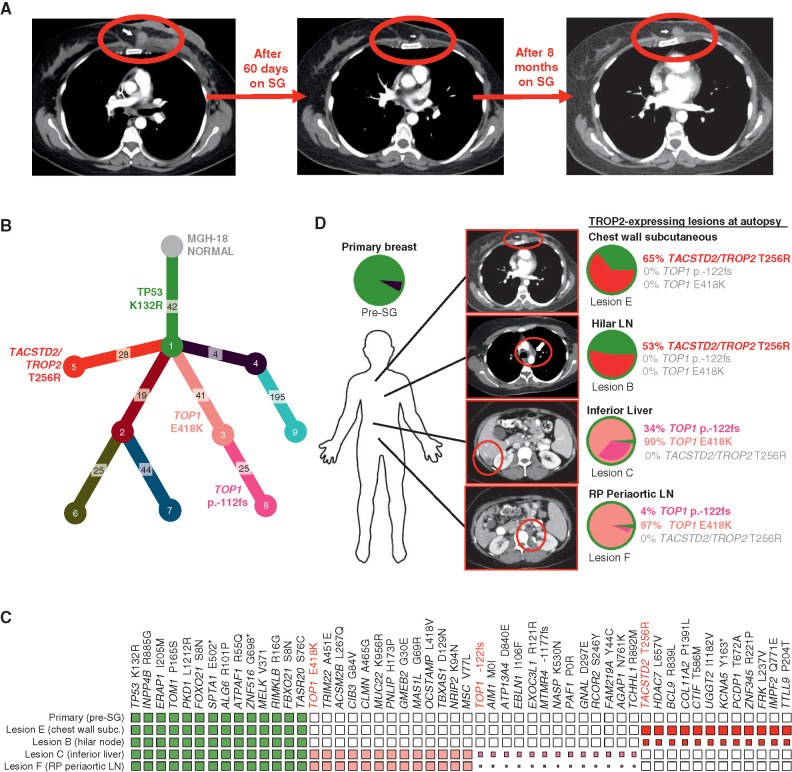
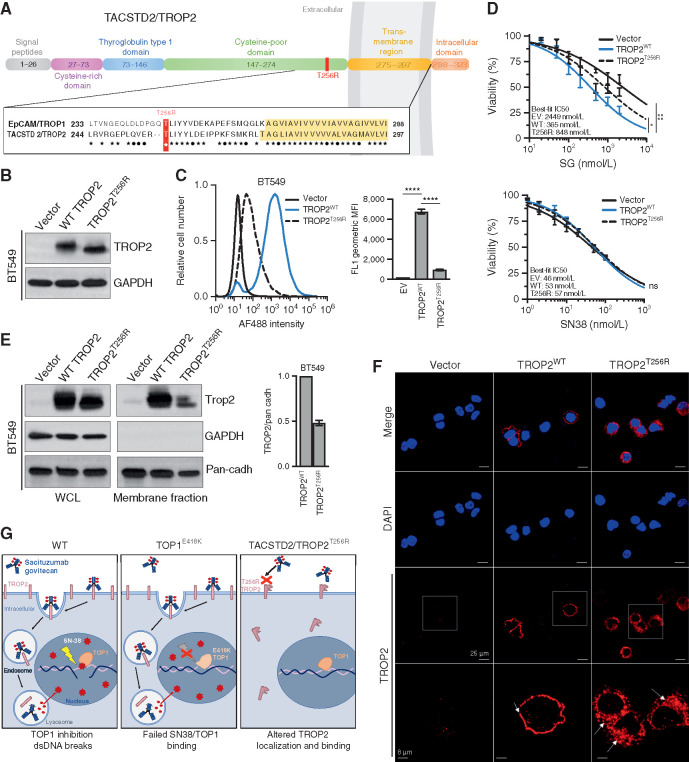
Sacituzumab govitecan (SG), the first antibody-drug conjugate (ADC) approved for triple-negative breast cancer, incorporates the anti-TROP2 antibody hRS7 conjugated to a topoisomerase-1 (TOP1) inhibitor payload. We sought to identify mechanisms of SG resistance through RNA and whole-exome sequencing of pretreatment and postprogression specimens. One patient exhibiting de novo progression lacked TROP2 expression, in contrast to robust TROP2 expression and focal genomic amplification of TACSTD2/TROP2 observed in a patient with a deep, prolonged response to SG. Analysis of acquired genomic resistance in this case revealed one phylogenetic branch harboring a canonical TOP1 E418K resistance mutation and subsequent frameshift TOP1 mutation, whereas a distinct branch exhibited a novel TACSTD2/TROP2 T256R missense mutation. Reconstitution experiments demonstrated that TROP2T256R confers SG resistance via defective plasma membrane localization and reduced cell-surface binding by hRS7. These findings highlight parallel genomic alterations in both antibody and payload targets associated with resistance to SG. SIGNIFICANCE: These findings underscore TROP2 as a response determinant and reveal acquired SG resistance mechanisms involving the direct antibody and drug payload targets in distinct metastatic subclones of an individual patient. This study highlights the specificity of SG and illustrates how such mechanisms will inform therapeutic strategies to overcome ADC resistance.This article is highlighted in the In This Issue feature, p. 2355.
©2021 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
S. Sun reports grants from NCI, Terri Brodeur Breast Cancer Foundation, and Massachusetts General Hospital during the conduct of the study. I. Leshchiner reports personal fees from PACT Pharma, Inc and Enov1, LLC outside the submitted work. A. Kambadakone reports grants from Philips Healthcare, GE Healthcare, PanCAN, personal fees from Bayer, and other support from Siemens Healthcare outside the submitted work. S.J. Isakoff reports personal fees from Immunomedics, Mylan, Myriad, Puma, Novartis, Seattle Genetics, grants and personal fees from OncoPep and AbbVie, grants from Merck, AstraZeneca, and Genentech outside the submitted work. D. Juric reports grants and personal fees from Novartis, Genentech, Syros, grants from Pfizer, Takeda, personal fees from Relay, PIC Therapeutics, Mapkure, Vibliome, grants and personal fees from Eisai, grants from InventisBio, Arvinas, Ribon Therapeutics, and Infinity Pharmaceuticals outside the submitted work. G. Getz reports grants from IBM during the conduct of the study; and receives research funds from Pharmacyclics. G. Getz is a founder, consultant, and holds privately held equity in Scorpion Therapeutics. A. Bardia reports grants from Genentech, Novartis, Pfizer, Merck, Sanofi, Radius Health, Immunomedics, and Diiachi Pharma/AstraZeneca, and personal fees from Pfizer, Novartis, Genentech, Merck, Radius Health, Immunomedics, Taiho, Sanofi, Diiachi Pharma/AstraZeneca, Puma; Biothernostics Inc., Phillips, Eli Lilly, Foundation Medicine, during the conduct of the study; personal fees from Pfizer, Novartis, Genentech, Merck, Radius Health, Immunomedics, Taiho, Sanofi, Diiachi Pharma/AstraZeneca, Puma; Biothernostics Inc., Phillips, Eli Lilly, Foundation Medicine, and grants from Genentech, Novartis, Pfizer, Merck, Sanofi, Radius Health, Immunomedics, and Diiachi Pharma/AstraZeneca outside the submitted work. L.W. Ellisen reports grants from DOD/CDMRP and other support from Tracey Davis Memorial Fund during the conduct of the study; and sacituzumab (drug only) provided by the sponsor for
Figures
References
-
- Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol 2010;7:683–92. - PubMed
-
- Gradishar WJ, Anderson BO, Abraham J, Aft R, Agnese D, Allison KH, et al. Breast cancer, version 3.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2020;18:452–78. - PubMed
-
- Nicolo E, Zagami P, Curigliano G. Antibody-drug conjugates in breast cancer: the chemotherapy of the future? Curr Opin Oncol 2020;32:494–502. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
