

CRISPRclassify: Repeat-Based Classification of CRISPR Loci
- PMID: 34406047
- PMCID: PMC8392126
- DOI: 10.1089/crispr.2021.0021
CRISPRclassify: Repeat-Based Classification of CRISPR Loci
Abstract
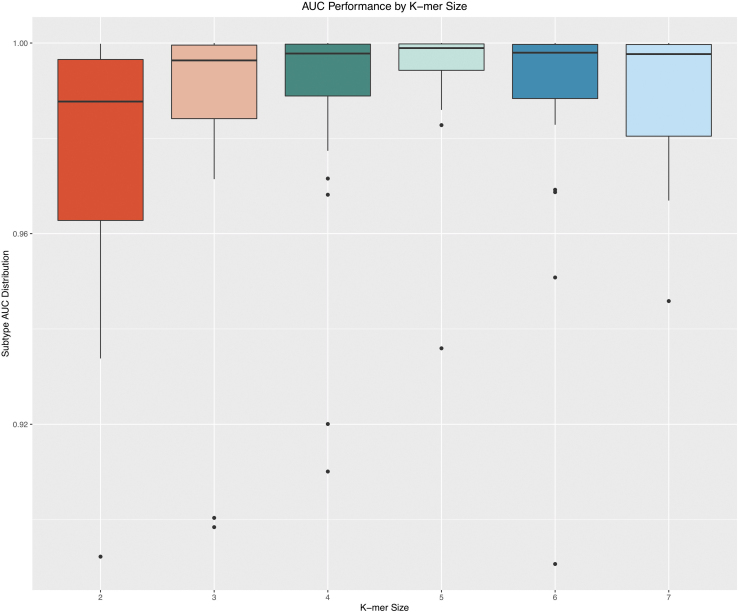
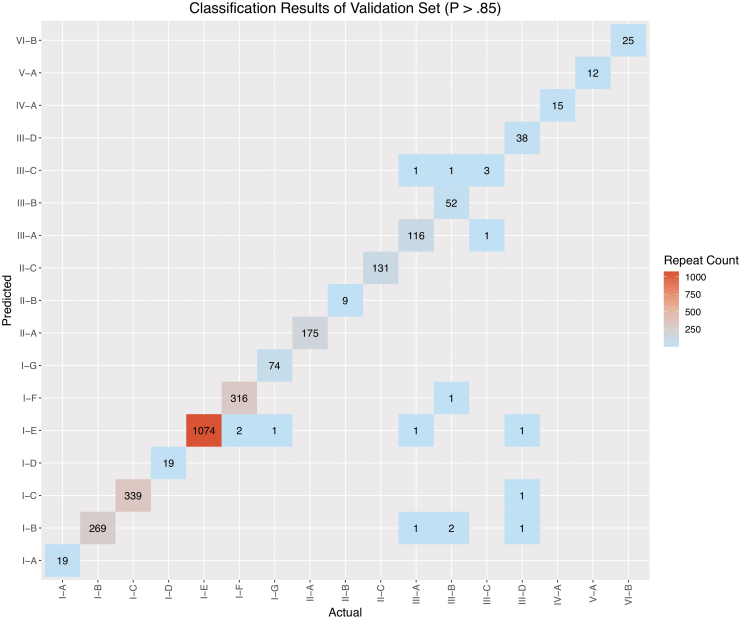
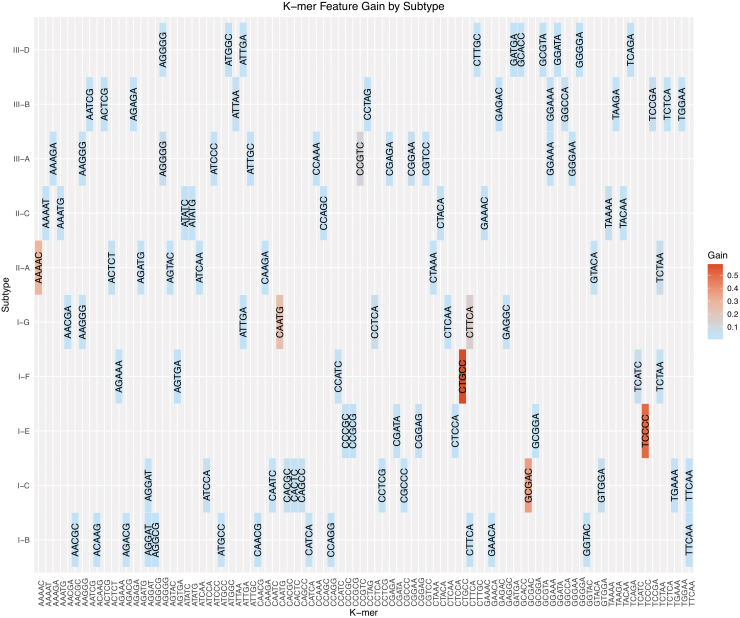
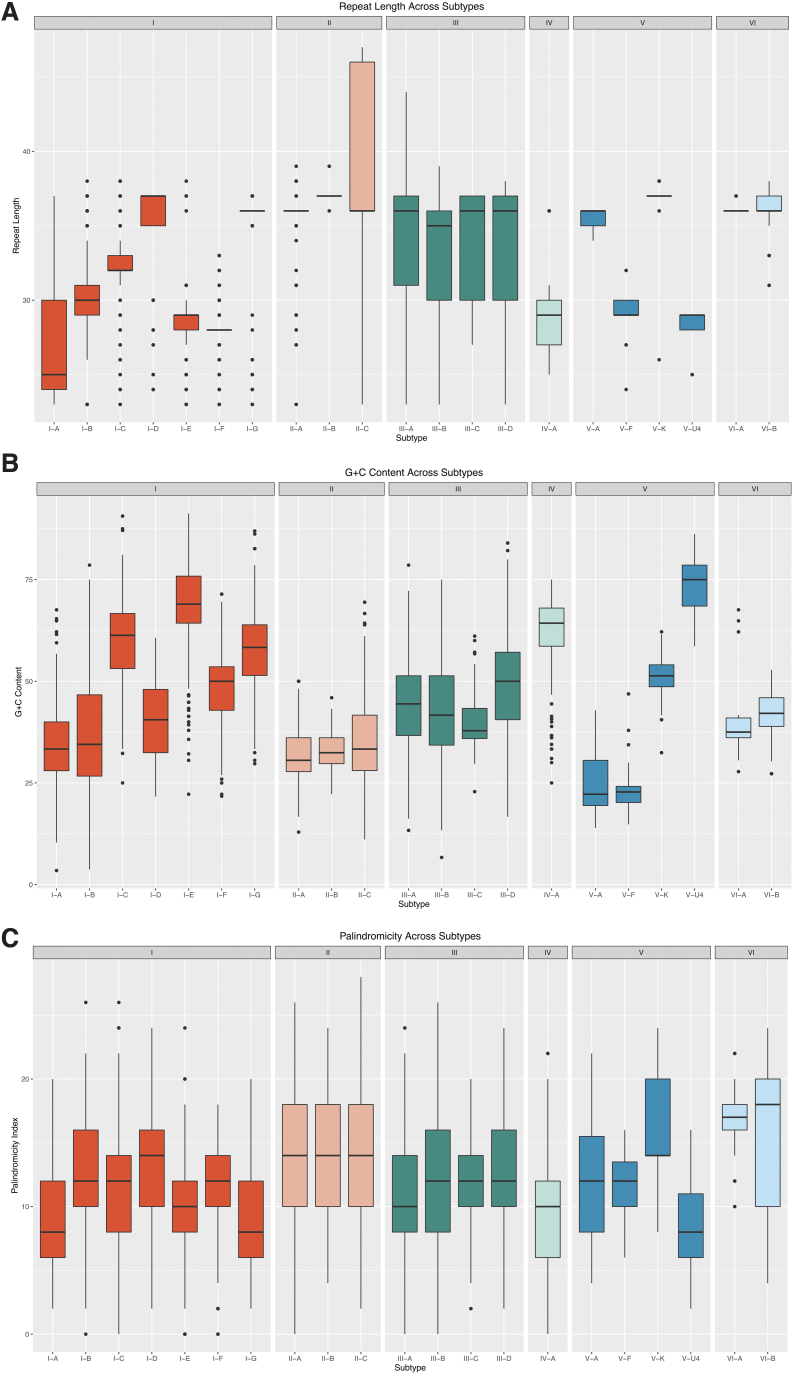
Detection and classification of CRISPR-Cas systems in metagenomic data have become increasingly prevalent in recent years due to their potential for diverse applications in genome editing. Traditionally, CRISPR-Cas systems are classified through reference-based identification of proximate cas genes. Here, we present a machine learning approach for the detection and classification of CRISPR loci using repeat sequences in a cas-independent context, enabling identification of unclassified loci missed by traditional cas-based approaches. Using biological attributes of the CRISPR repeat, the core element in CRISPR arrays, and leveraging methods from natural language processing, we developed a machine learning model capable of accurate classification of CRISPR loci in an extensive set of metagenomes, resulting in an F1 measure of 0.82 across all predictions and an F1 measure of 0.97 when limiting to classifications with probabilities >0.85. Furthermore, assessing performance on novel repeats yielded an F1 measure of 0.96. Although the performance of cas-based identification will exceed that of a repeat-based approach in many cases, CRISPRclassify provides an efficient approach to classification of CRISPR loci for cases in which cas gene information is unavailable, such as metagenomes and fragmented genome assemblies.
Conflict of interest statement
The authors declare no potential conflict of interest.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
