

Quantifying the relationship between genetic diversity and population size suggests natural selection cannot explain Lewontin's Paradox
- PMID: 34409937
- PMCID: PMC8486380
- DOI: 10.7554/eLife.67509
Quantifying the relationship between genetic diversity and population size suggests natural selection cannot explain Lewontin's Paradox
Abstract
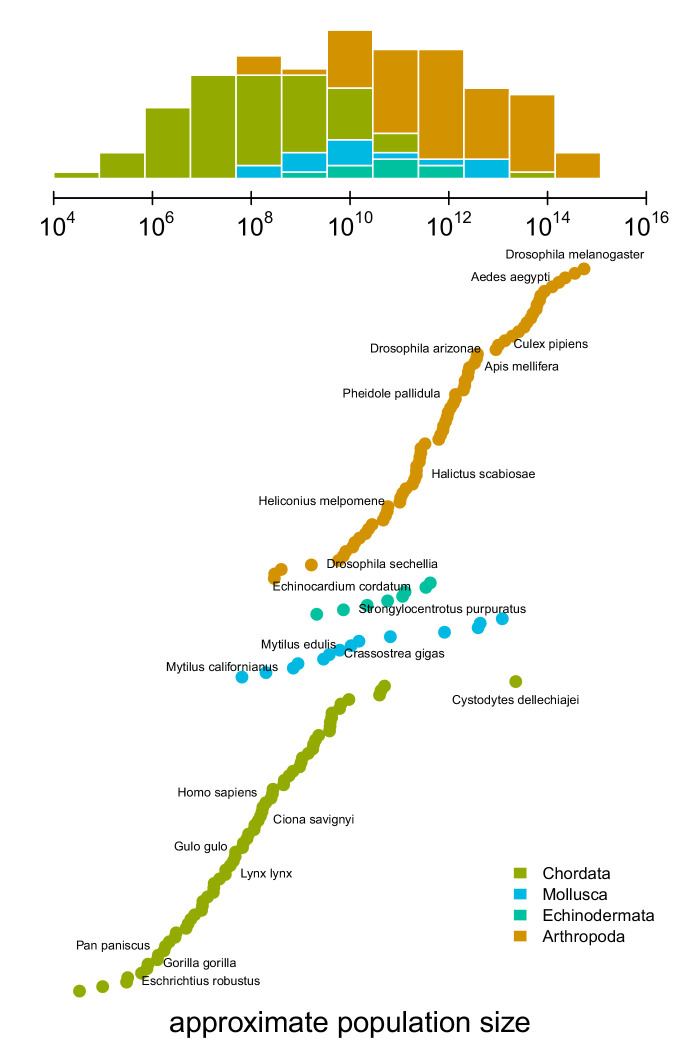
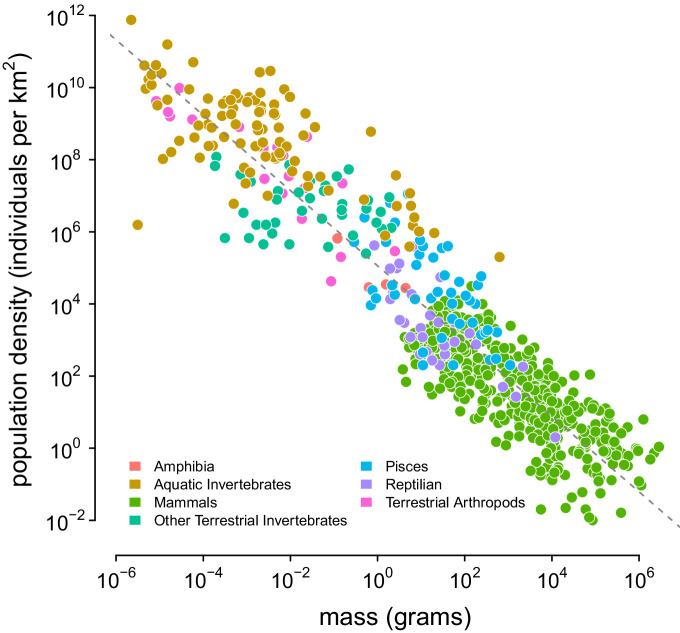
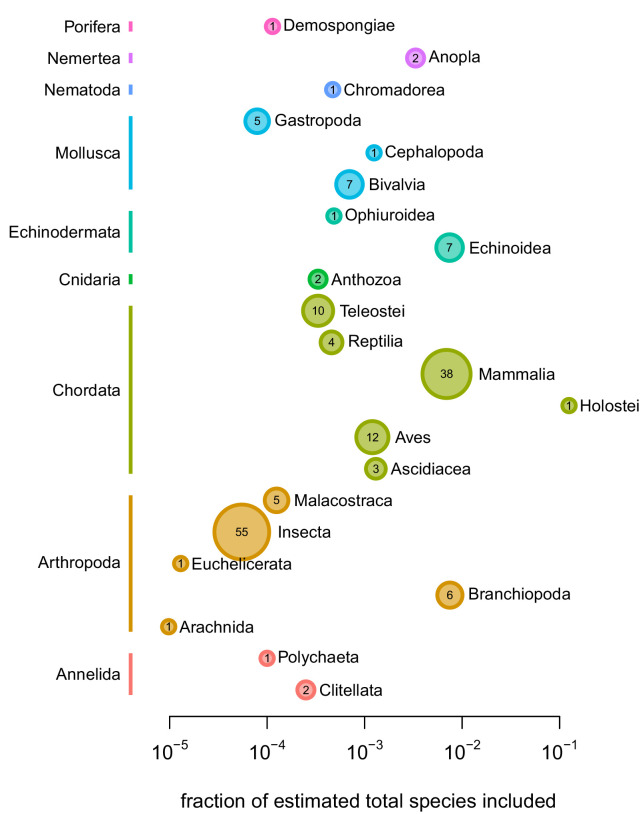
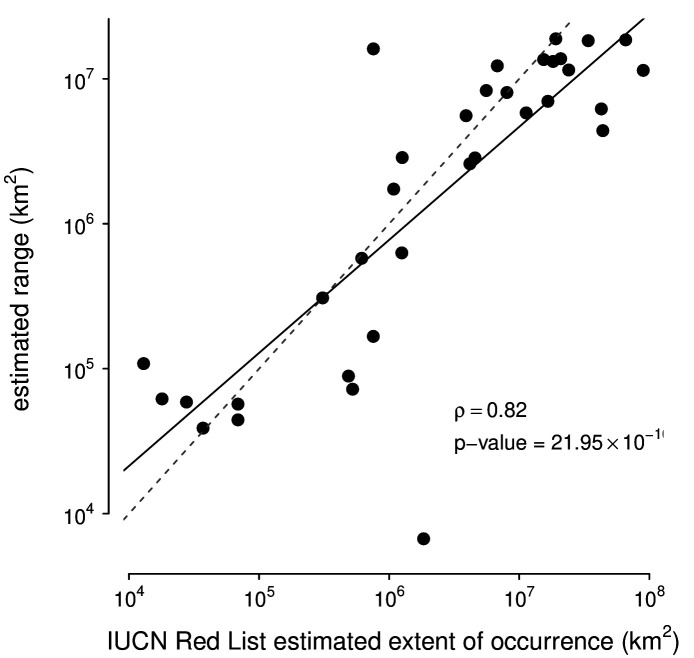
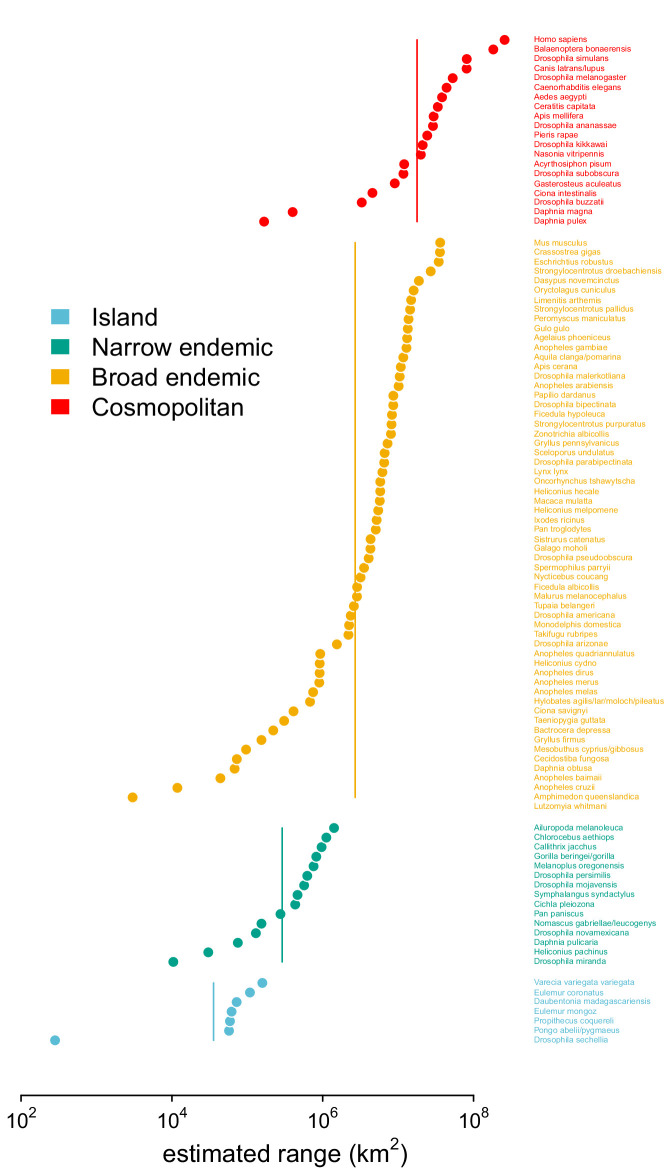
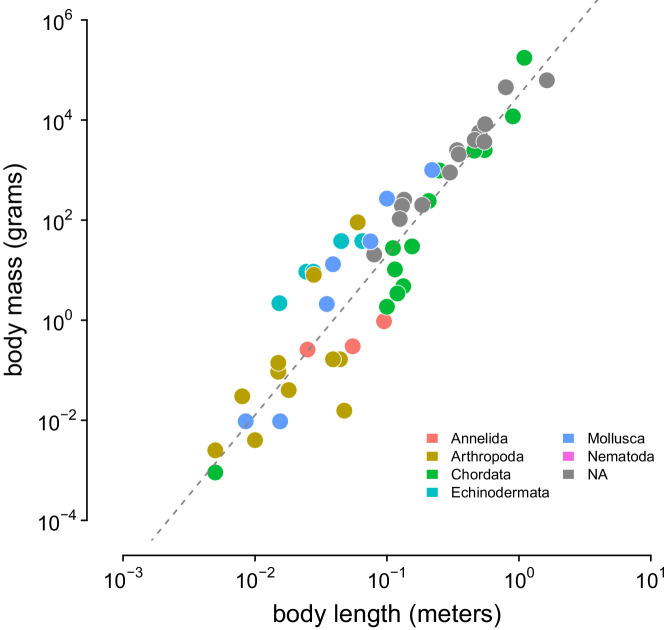
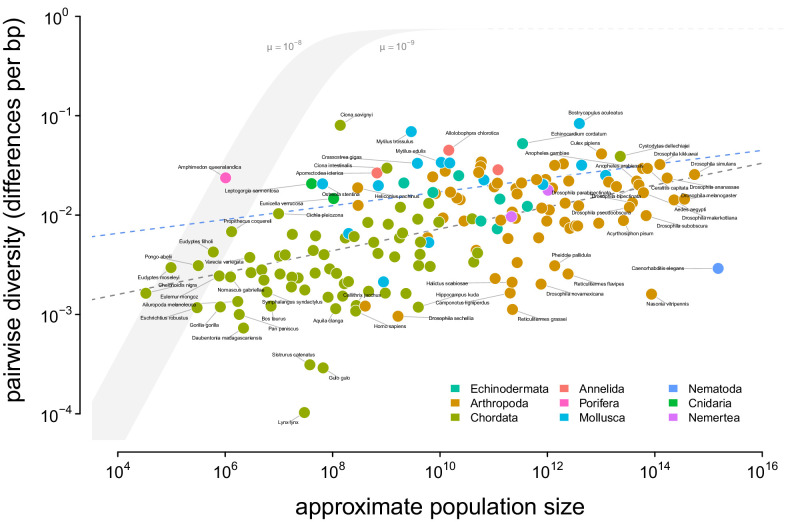
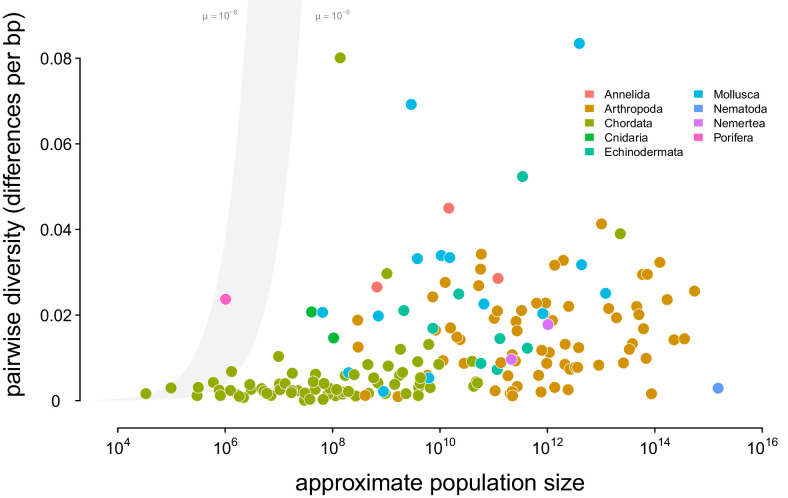
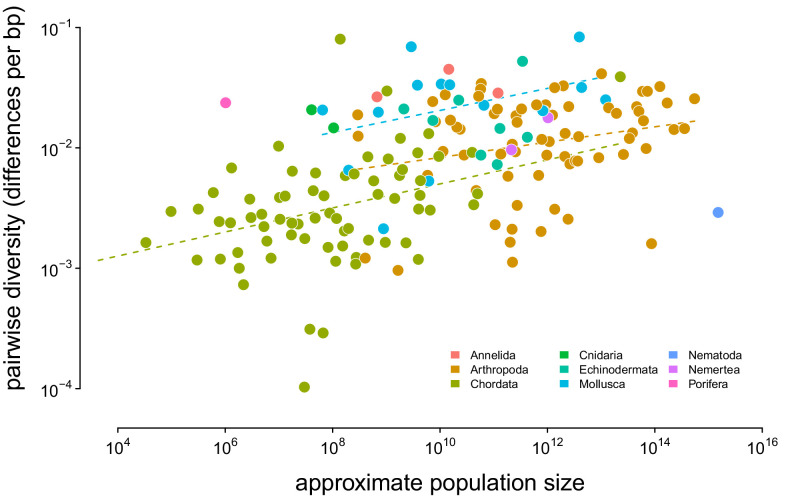
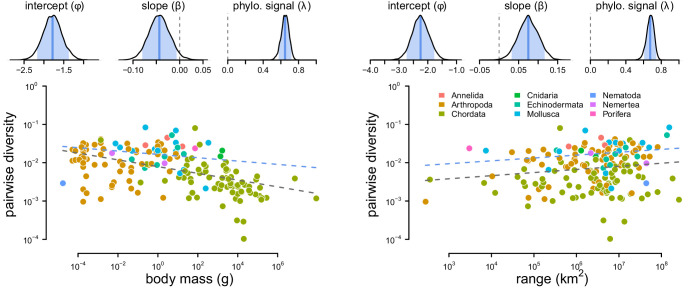
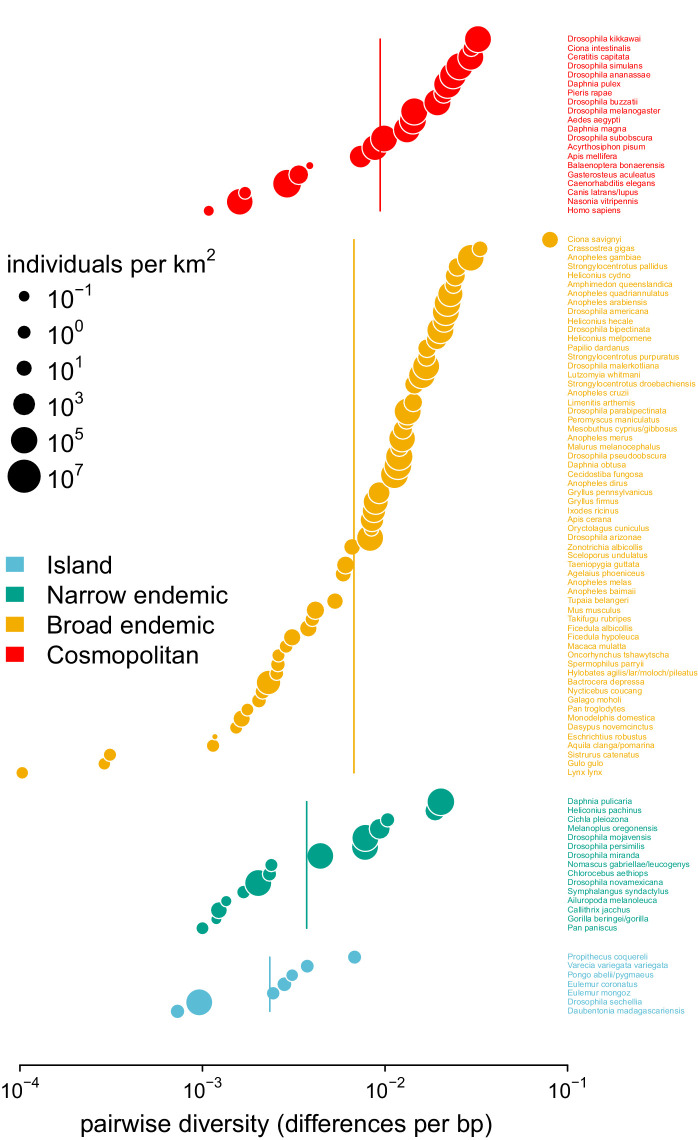
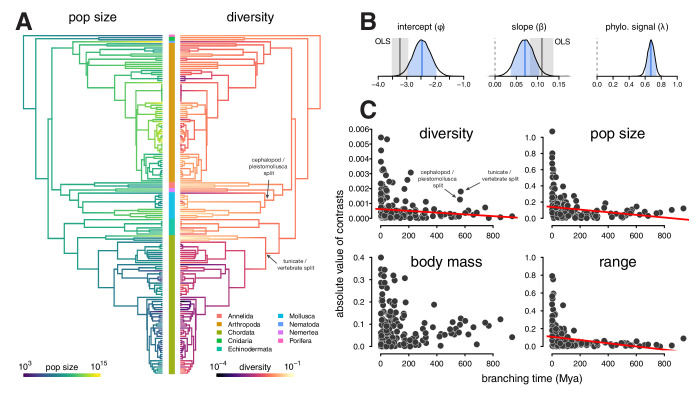
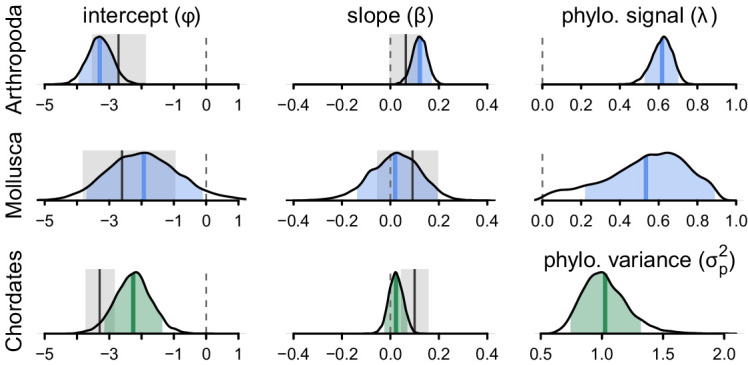
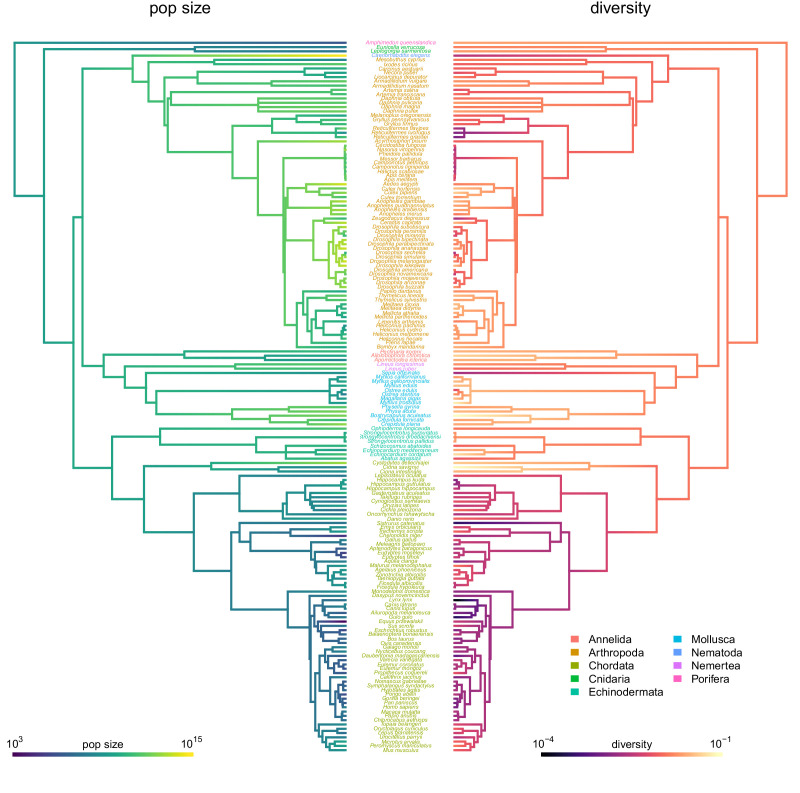
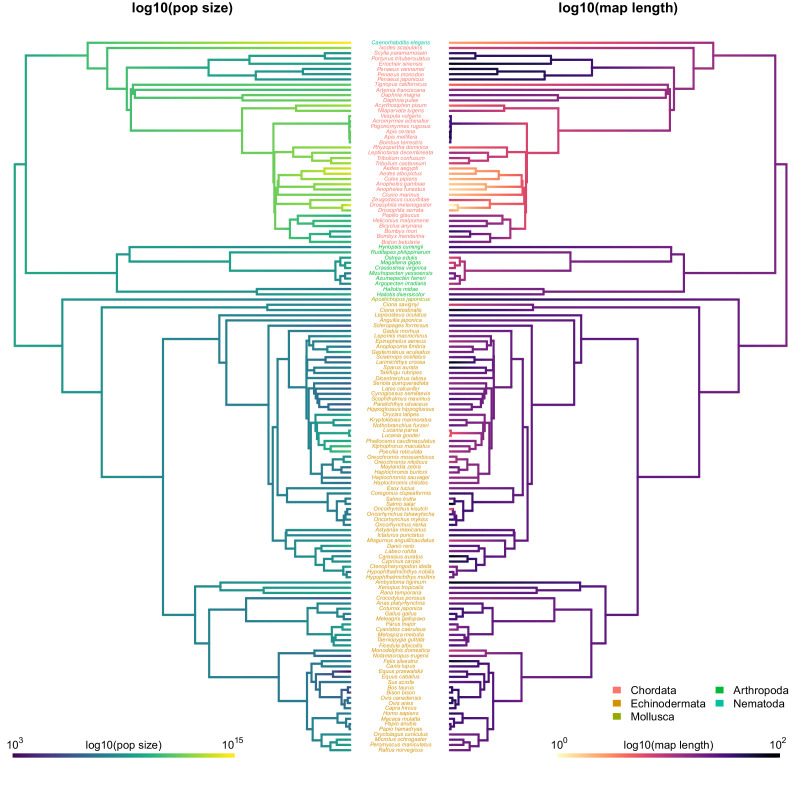
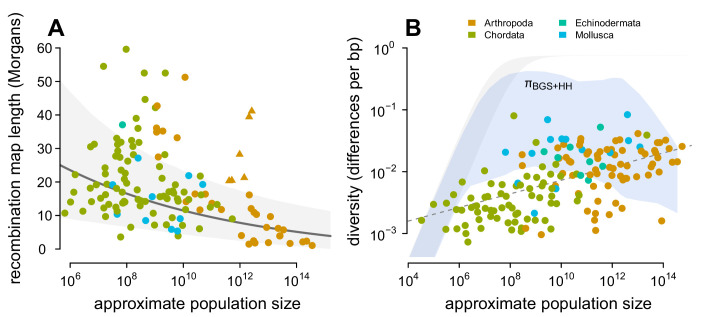
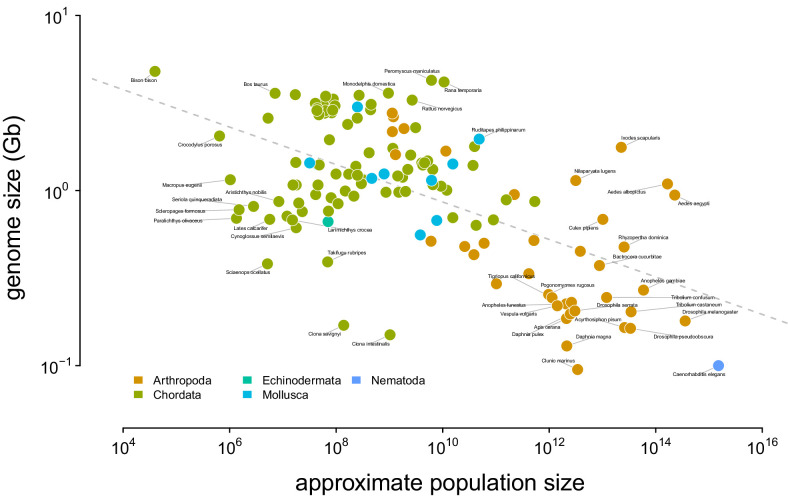
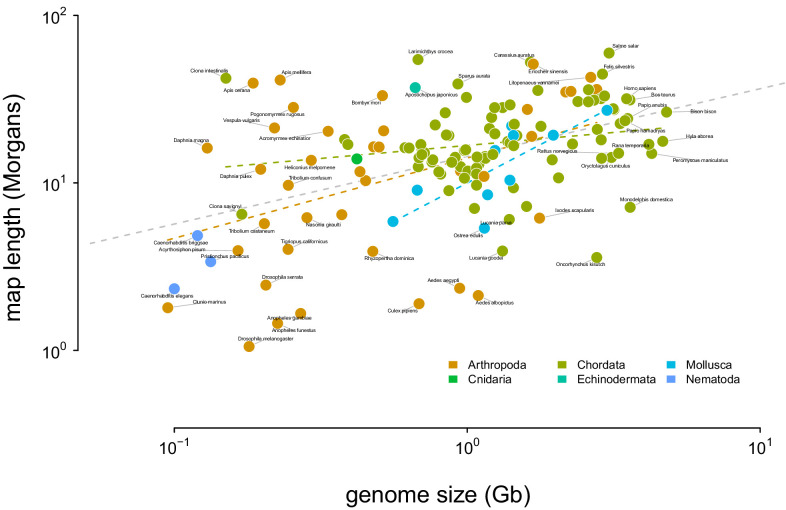
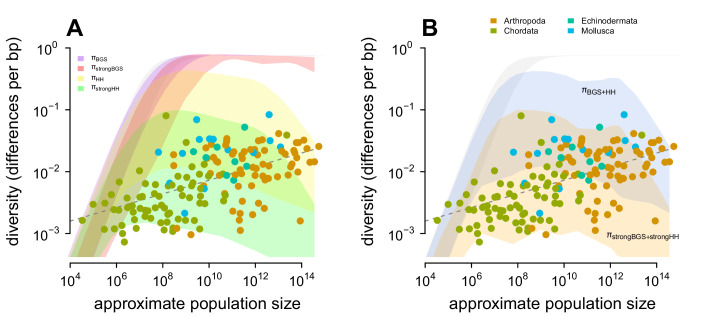
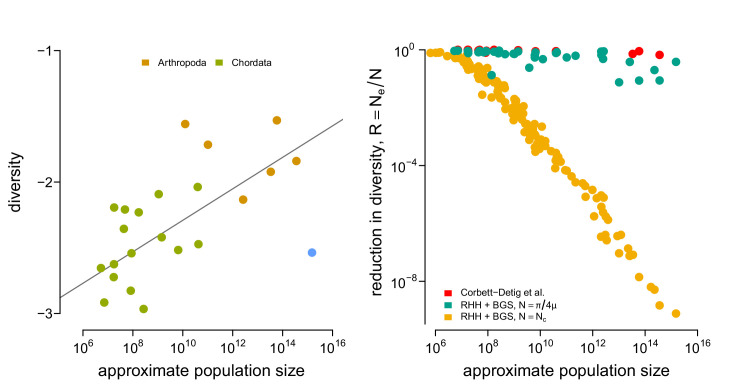
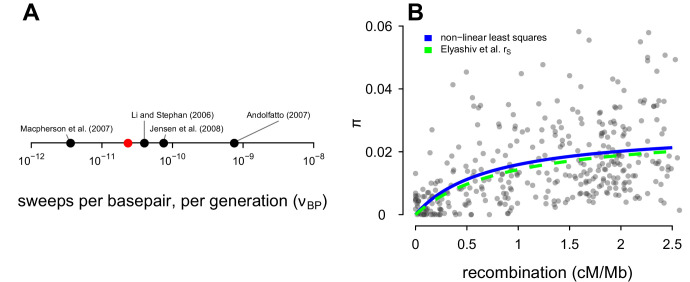
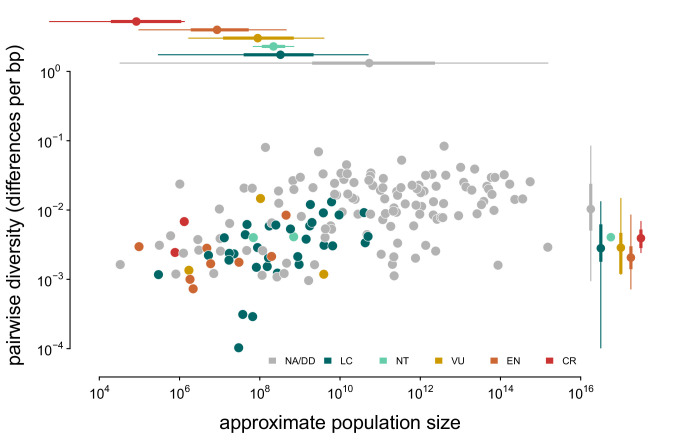
Neutral theory predicts that genetic diversity increases with population size, yet observed levels of diversity across metazoans vary only two orders of magnitude while population sizes vary over several. This unexpectedly narrow range of diversity is known as Lewontin's Paradox of Variation (1974). While some have suggested selection constrains diversity, tests of this hypothesis seem to fall short. Here, I revisit Lewontin's Paradox to assess whether current models of linked selection are capable of reducing diversity to this extent. To quantify the discrepancy between pairwise diversity and census population sizes across species, I combine previously-published estimates of pairwise diversity from 172 metazoan taxa with newly derived estimates of census sizes. Using phylogenetic comparative methods, I show this relationship is significant accounting for phylogeny, but with high phylogenetic signal and evidence that some lineages experience shifts in the evolutionary rate of diversity deep in the past. Additionally, I find a negative relationship between recombination map length and census size, suggesting abundant species have less recombination and experience greater reductions in diversity due to linked selection. However, I show that even assuming strong and abundant selection, models of linked selection are unlikely to explain the observed relationship between diversity and census sizes across species.
Keywords: Lewontin's Paradox; evolutionary biology; linked selection; none; phylogenetic comparative methods.
© 2021, Buffalo.
Conflict of interest statement
VB No competing interests declared
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
