

Easy-Prime: a machine learning-based prime editor design tool
- PMID: 34412673
- PMCID: PMC8377858
- DOI: 10.1186/s13059-021-02458-0
Easy-Prime: a machine learning-based prime editor design tool
Abstract
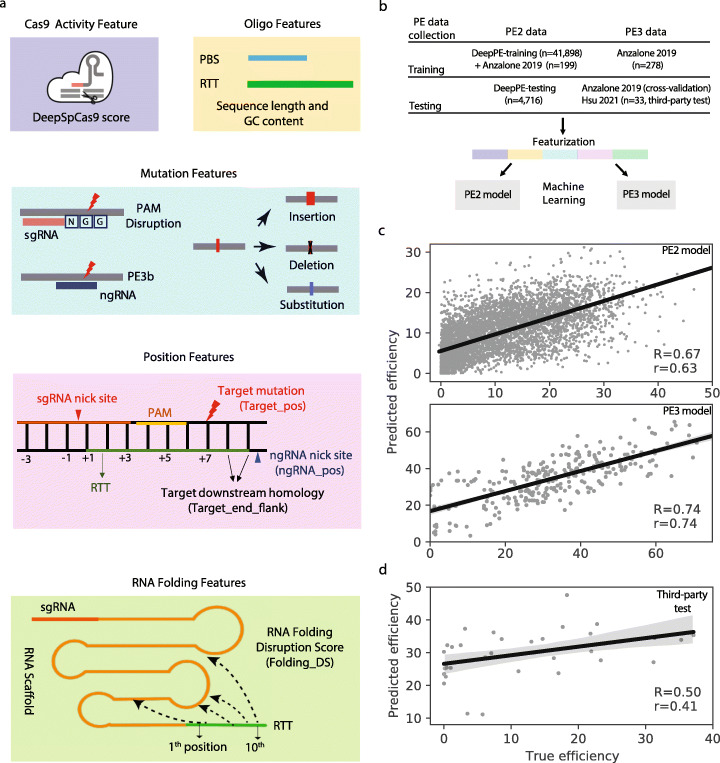
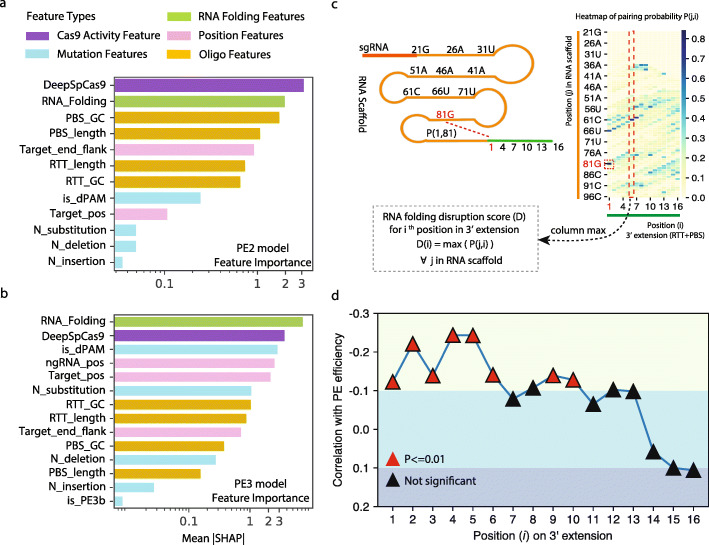
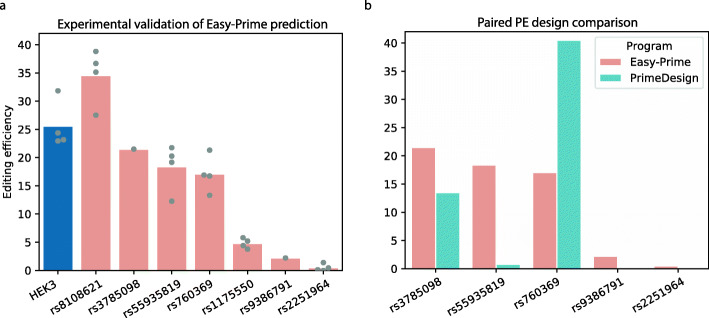
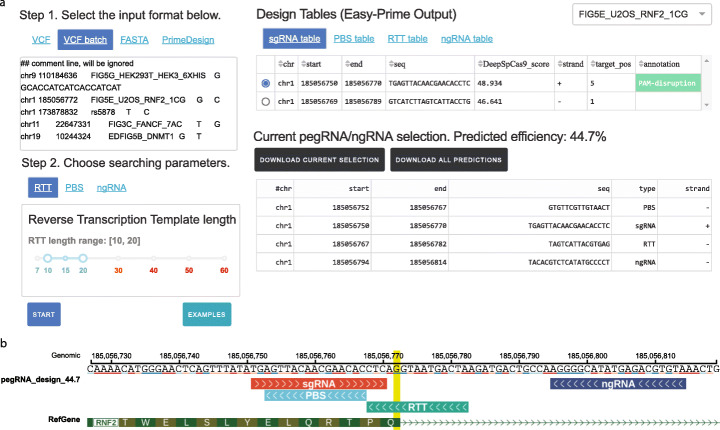
Prime editing is a revolutionary genome-editing technology that can make a wide range of precise edits in DNA. However, designing highly efficient prime editors (PEs) remains challenging. We develop Easy-Prime, a machine learning-based program trained with multiple published data sources. Easy-Prime captures both known and novel features, such as RNA folding structure, and optimizes feature combinations to improve editing efficiency. We provide optimized PE design for installation of 89.5% of 152,351 GWAS variants. Easy-Prime is available both as a command line tool and an interactive PE design server at: http://easy-prime.cc/ .
Keywords: Machine learning; Prime editor; pegRNA design.
© 2021. The Author(s).
Conflict of interest statement
S.Q.T. is a member of the scientific advisory board of Kromatid and Twelve Bio.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
