

IMProv: A Resource for Cross-link-Driven Structure Modeling that Accommodates Protein Dynamics
- PMID: 34418567
- PMCID: PMC8452774
- DOI: 10.1016/j.mcpro.2021.100139
IMProv: A Resource for Cross-link-Driven Structure Modeling that Accommodates Protein Dynamics
Abstract
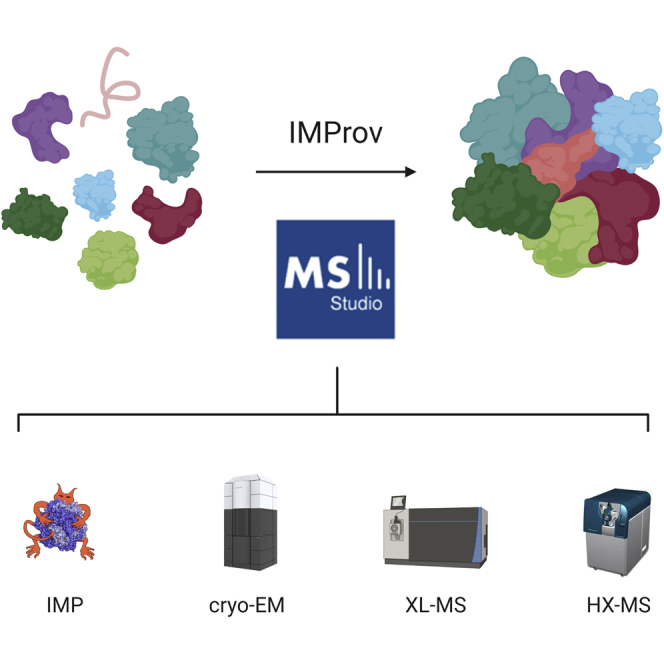
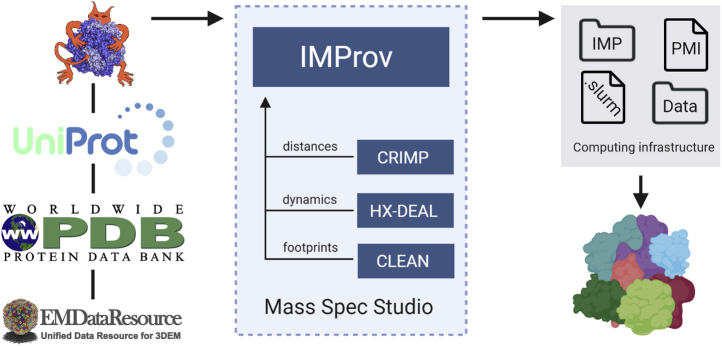
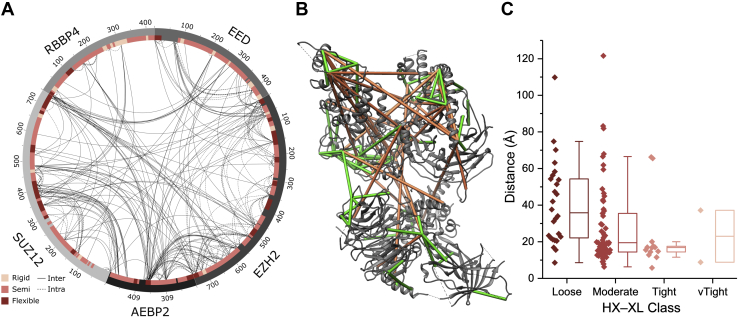
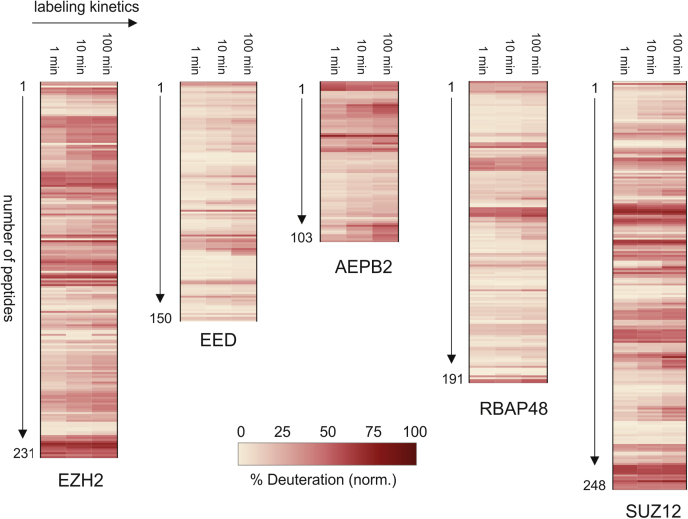
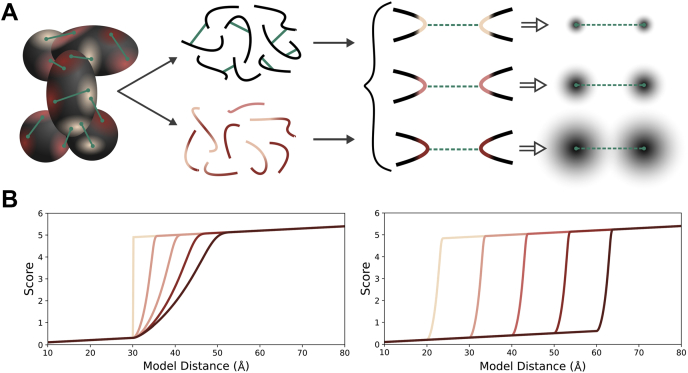
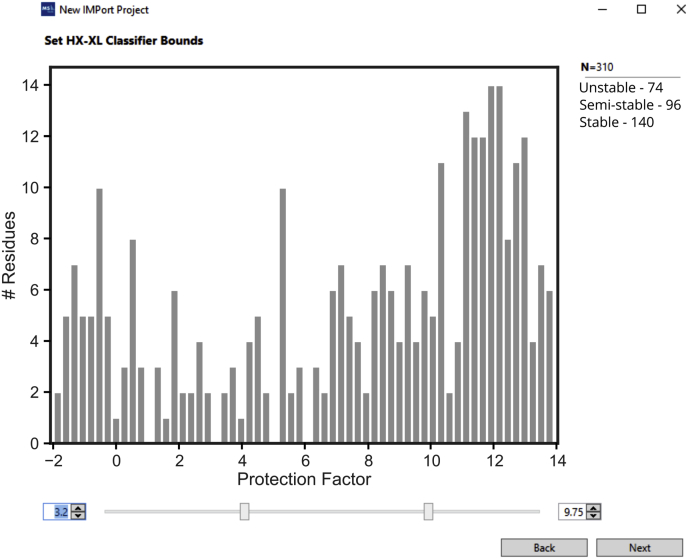
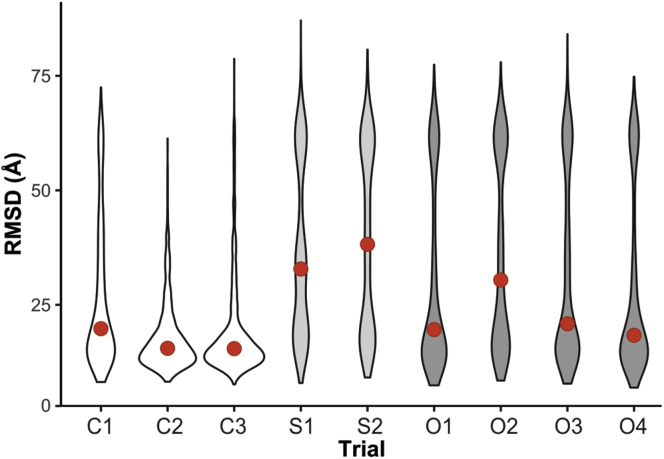
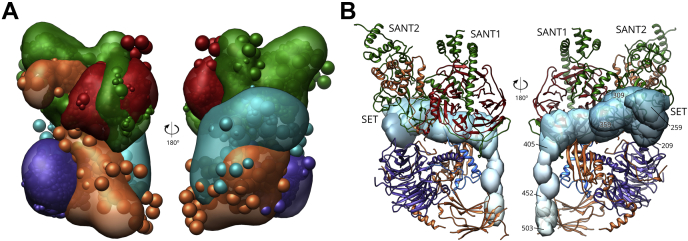
Proteomics methodology has expanded to include protein structural analysis, primarily through cross-linking mass spectrometry (XL-MS) and hydrogen-deuterium exchange mass spectrometry (HX-MS). However, while the structural proteomics community has effective tools for primary data analysis, there is a need for structure modeling pipelines that are accessible to the proteomics specialist. Integrative structural biology requires the aggregation of multiple distinct types of data to generate models that satisfy all inputs. Here, we describe IMProv, an app in the Mass Spec Studio that combines XL-MS data with other structural data, such as cryo-EM densities and crystallographic structures, for integrative structure modeling on high-performance computing platforms. The resource provides an easily deployed bundle that includes the open-source Integrative Modeling Platform program (IMP) and its dependencies. IMProv also provides functionality to adjust cross-link distance restraints according to the underlying dynamics of cross-linked sites, as characterized by HX-MS. A dynamics-driven conditioning of restraint values can improve structure modeling precision, as illustrated by an integrative structure of the five-membered Polycomb Repressive Complex 2. IMProv is extensible to additional types of data.
Keywords: Polycomb Repressive Complex 2; crosslinking; cryo-electron microscopy; hydrogen-deuterium exchange; integrative modeling; structural mass spectrometry.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
References
-
- Kastritis P.L., Reilly F.J.O., Bock T., Li Y., Rogon M.Z., Buczak K., Romanov N., Betts M.J., Bui K.H., Hagen W.J., Hennrich L., Mackmull M., Rappsilber J., Russell R.B., Bork P. Capturing protein communities by structural proteomics in a thermophilic eukaryote. Mol. Syst. Biol. 2017;13:936. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
