

Mitochondrial Reactive Oxygen Species Are Essential for the Development of Psoriatic Inflammation
- PMID: 34421919
- PMCID: PMC8378889
- DOI: 10.3389/fimmu.2021.714897
Mitochondrial Reactive Oxygen Species Are Essential for the Development of Psoriatic Inflammation
Abstract
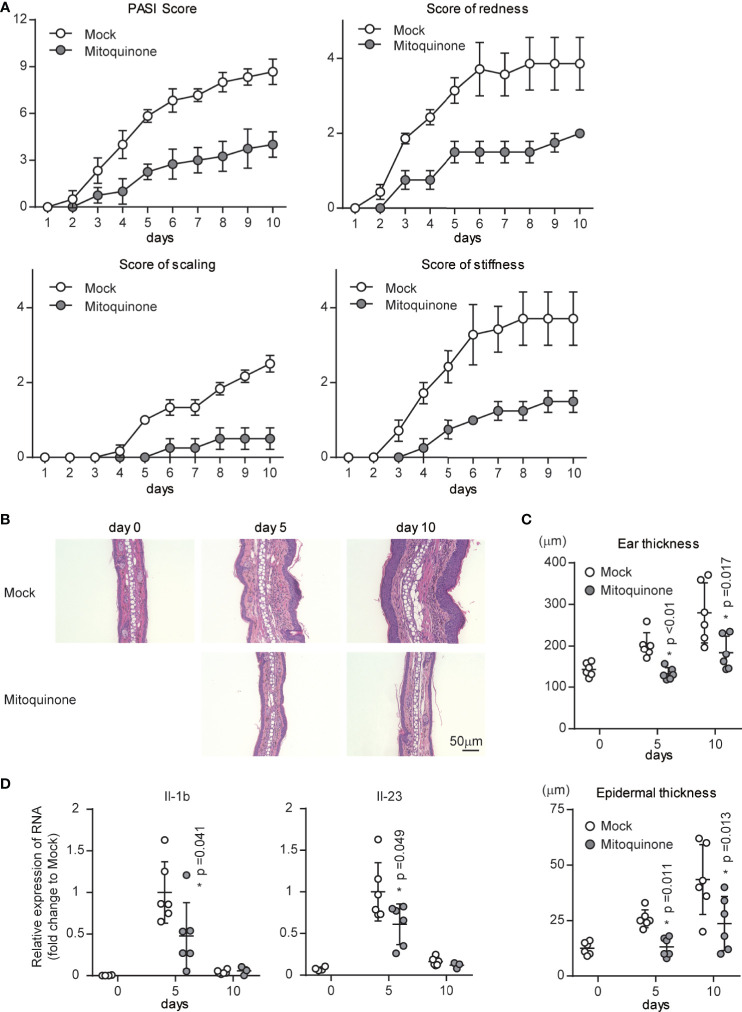
Psoriasis is a common immune-mediated, chronic, inflammatory skin disease that affects approximately 2-3% of the population worldwide. Although there is increasing evidence regarding the essential roles of the interleukin (IL)-23/IL-17 axis and dendritic cell (DC)-T cell crosstalk in the development of skin inflammation, the contributions of mitochondrial function to psoriasis are unclear. In a mouse model of imiquimod (IMQ)-induced psoriasiform skin inflammation, we found that hematopoietic cell-specific genetic deletion of p32/C1qbp, a regulator of mitochondrial protein synthesis and metabolism, protects mice from IMQ-induced psoriatic inflammation. Additionally, we demonstrate that p32/C1qbp is an important regulator of IMQ-induced DC activation, both in vivo and in vitro. We also found that p32/C1qbp-deficient DCs exhibited impaired production of IL-1β, IL-23, and mitochondrial reactive oxygen species (mtROS) after IMQ stimulation. Because the inhibition of mtROS suppressed IMQ-induced DC activation and psoriatic inflammation, we presume that p32/C1qbp and mtROS can serve as therapeutic targets in psoriasis.
Keywords: C1qbp/p32; IL-1β; dendritic cells; mitochondrial ROS; psoriasis.
Copyright © 2021 Mizuguchi, Gotoh, Nakashima, Setoyama, Takata, Ohga and Kang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous