

Inference of Nipah virus evolution, 1999-2015
- PMID: 34422315
- PMCID: PMC7947586
- DOI: 10.1093/ve/veaa062
Inference of Nipah virus evolution, 1999-2015
Abstract
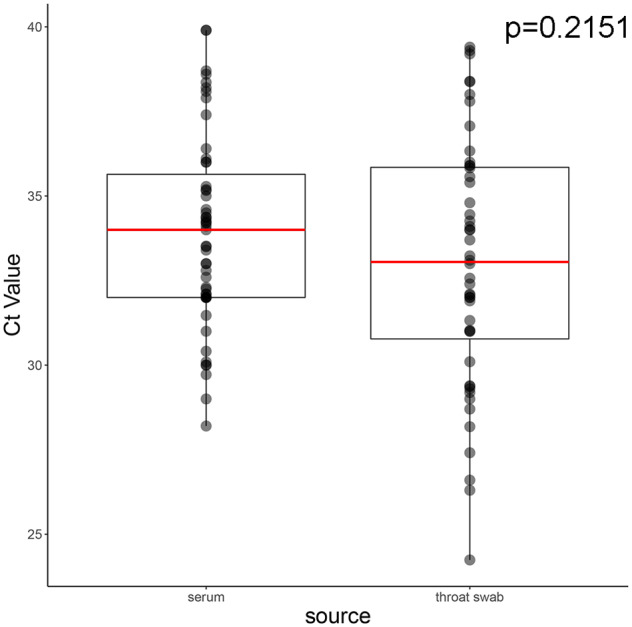
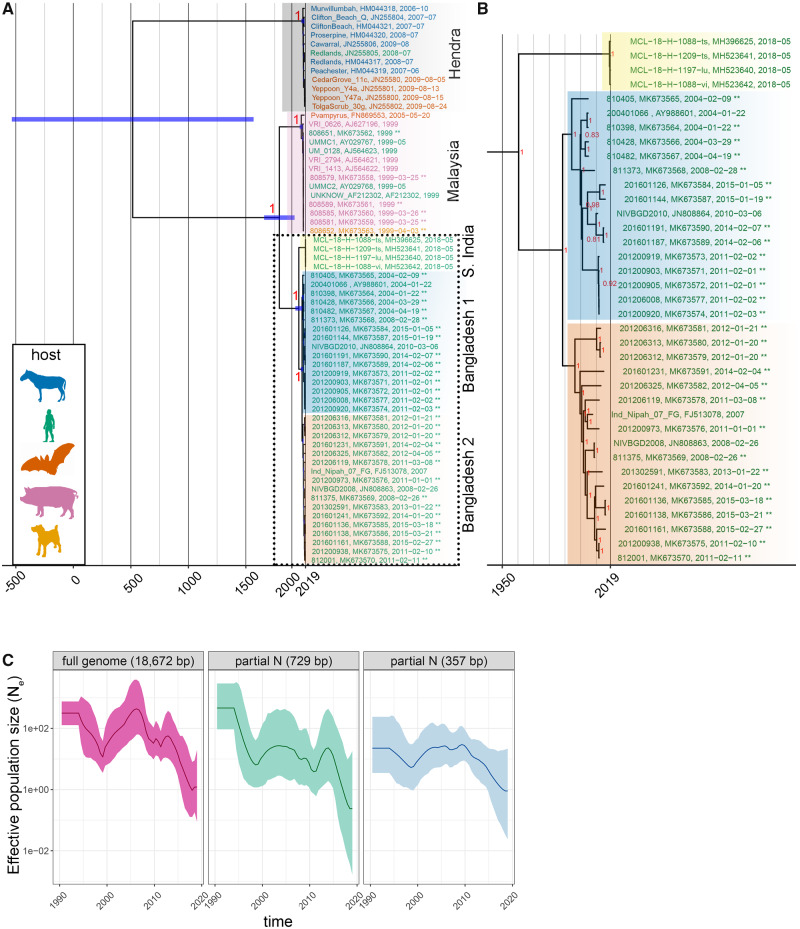
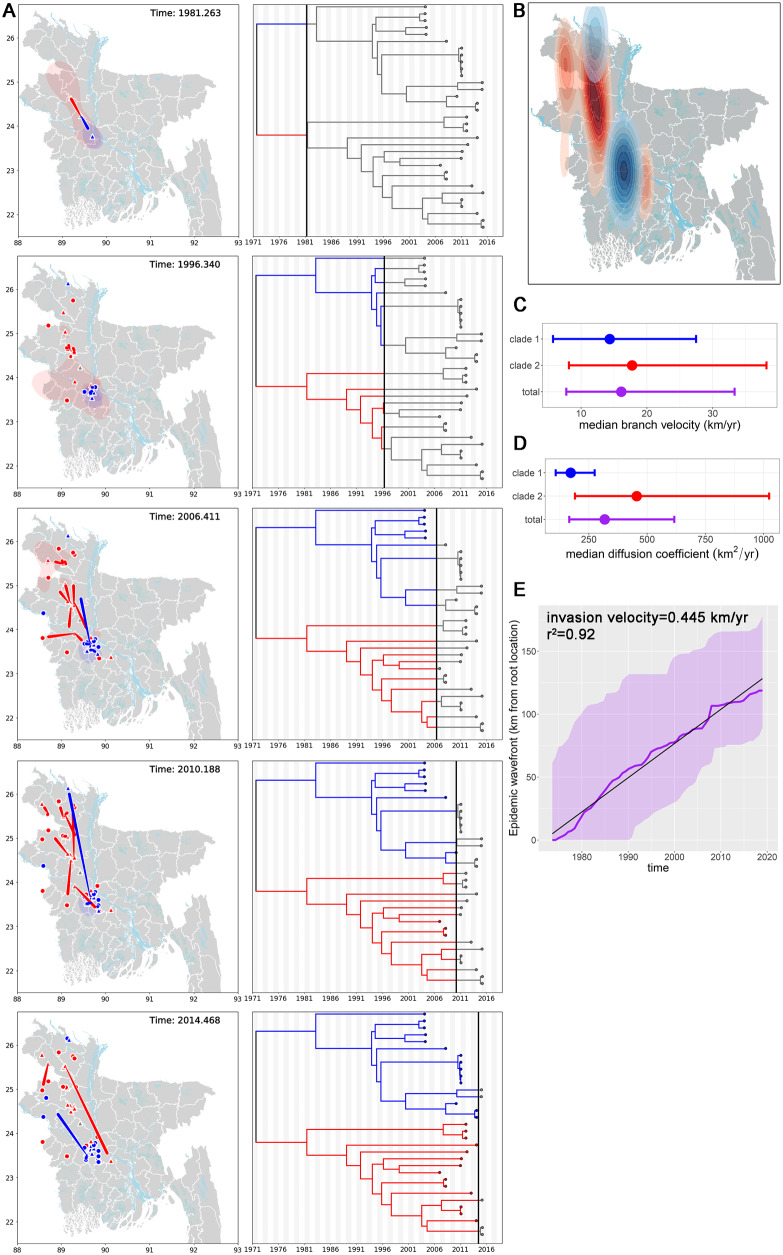
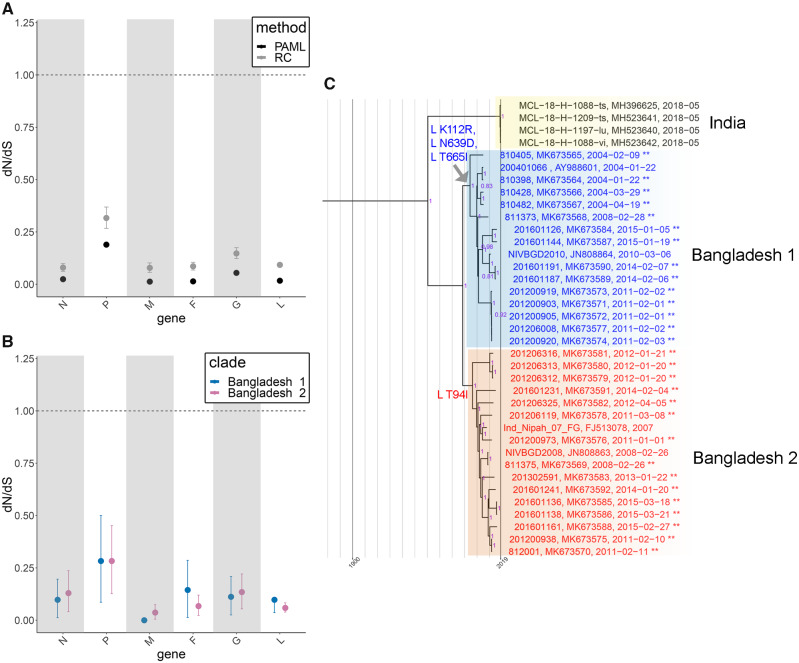
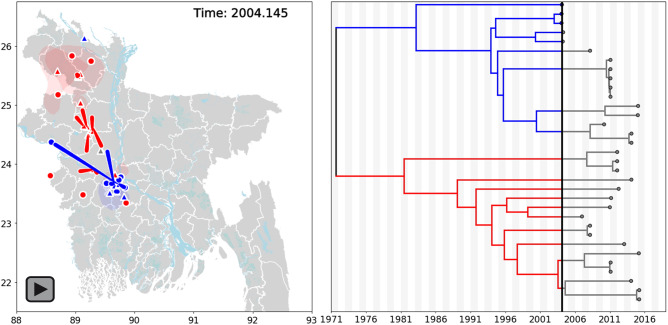
Despite near-annual human outbreaks of Nipah virus (NiV) disease in Bangladesh, typically due to individual spillover events from the local bat population, only twenty whole-genome NiV sequences exist from humans and ten from bats. NiV whole-genome sequences from annual outbreaks have been challenging to generate, primarily due to the low viral load in human throat swab and serum specimens. Here, we used targeted enrichment with custom NiV-specific probes and generated thirty-five additional unique full-length genomic sequences directly from human specimens and viral isolates. We inferred the temporal and geographic evolutionary history of NiV in Bangladesh and expanded a tool to visualize NiV spatio-temporal spread from a Bayesian continuous diffusion analysis. We observed that strains from Bangladesh segregated into two distinct clades that have intermingled geographically in Bangladesh over time and space. As these clades expanded geographically and temporally, we did not observe evidence for significant branch and site-specific selection, except for a single site in the Henipavirus L polymerase. However, the Bangladesh 1 and 2 clades are differentiated by mutations initially occurring in the polymerase, with additional mutations accumulating in the N, G, F, P, and L genes on external branches. Modeling the historic geographical and temporal spread demonstrates that while widespread, NiV does not exhibit significant genetic variation in Bangladesh. Thus, future public health measures should address whether NiV within in the bat population also exhibits comparable genetic variation, if zoonotic transmission results in a genetic bottleneck and if surveillance techniques are detecting only a subset of NiV.
Keywords: Nipah virus; phylogeography; selective pressure; virus evolution.
Published by Oxford University Press 2020. This work is written by a US Government employee and is in the public domain in the US.
Figures
References
-
- Arunkumar G. et al. (2019) ‘Outbreak Investigation of Nipah Virus Disease in Kerala, India, 2018’, The Journal of Infectious Diseases, 219: 1867–78. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
