

Comprehensive Wet-Bench and Bioinformatics Workflow for Complex Microbiota Using Oxford Nanopore Technologies
- PMID: 34427527
- PMCID: PMC8407471
- DOI: 10.1128/mSystems.00750-21
Comprehensive Wet-Bench and Bioinformatics Workflow for Complex Microbiota Using Oxford Nanopore Technologies
Abstract
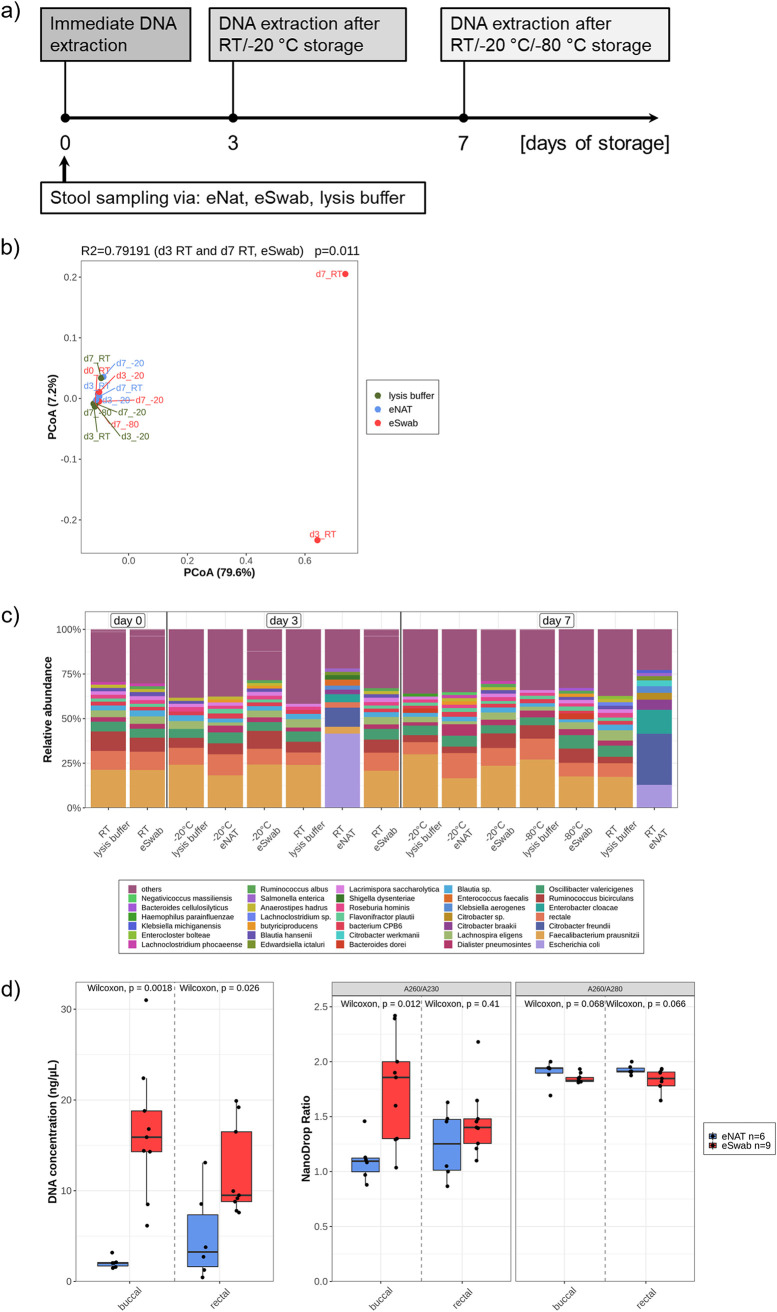
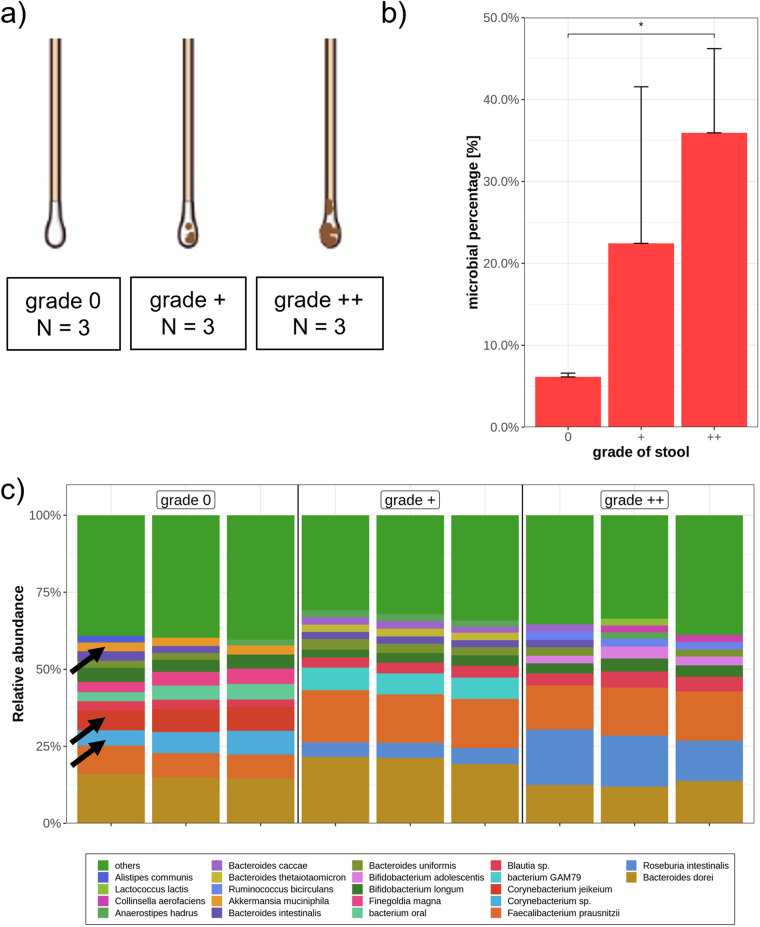
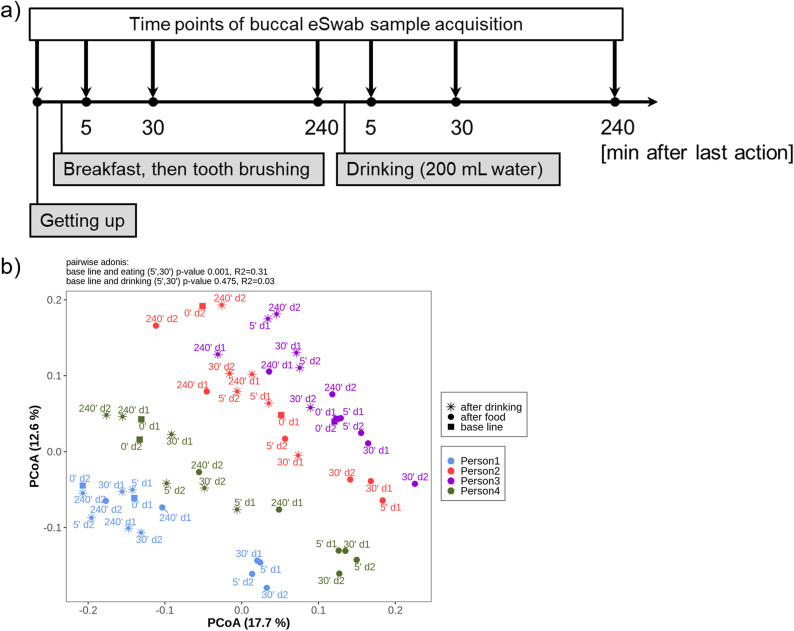
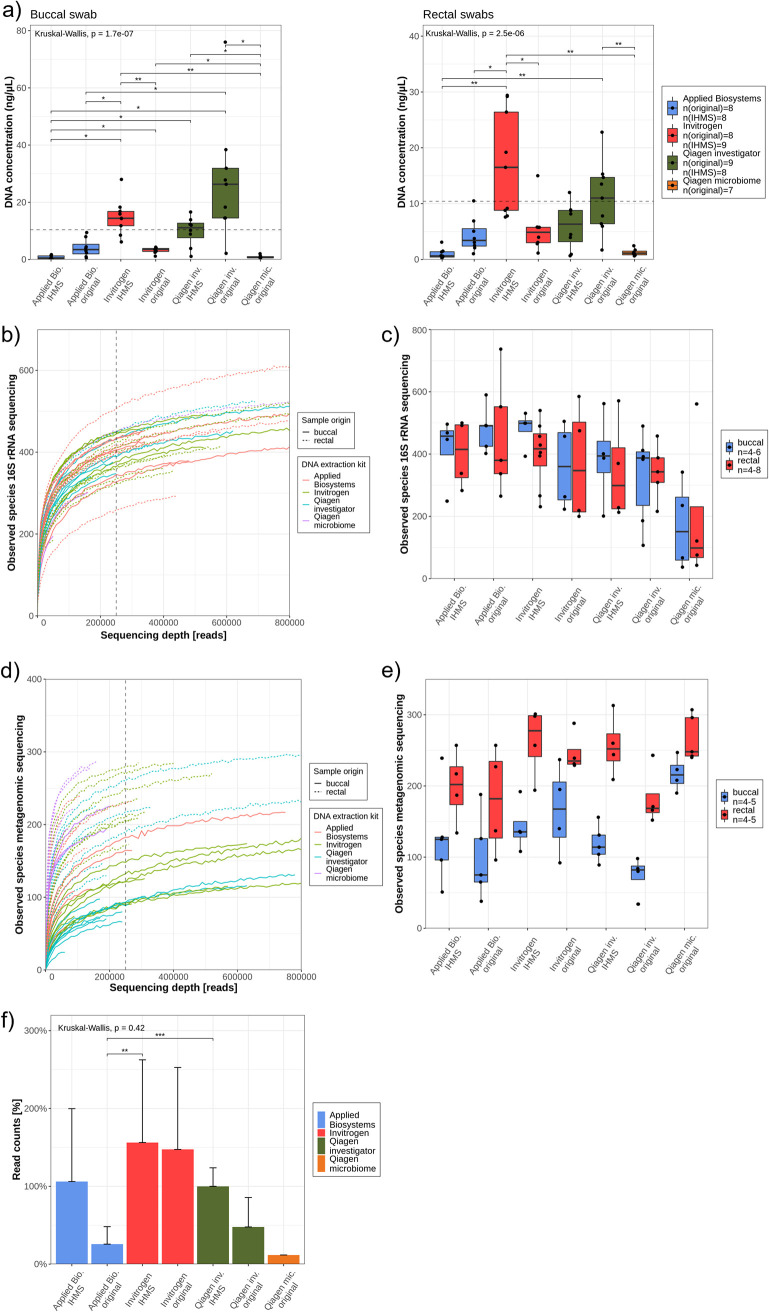
The advent of high-throughput sequencing techniques has recently provided an astonishing insight into the composition and function of the human microbiome. Next-generation sequencing (NGS) has become the gold standard for advanced microbiome analysis; however, 3rd generation real-time sequencing, such as Oxford Nanopore Technologies (ONT), enables rapid sequencing from several kilobases to >2 Mb with high resolution. Despite the wide availability and the enormous potential for clinical and translational applications, ONT is poorly standardized in terms of sampling and storage conditions, DNA extraction, library creation, and bioinformatic classification. Here, we present a comprehensive analysis pipeline with sampling, storage, DNA extraction, library preparation, and bioinformatic evaluation for complex microbiomes sequenced with ONT. Our findings from buccal and rectal swabs and DNA extraction experiments indicate that methods that were approved for NGS microbiome analysis cannot be simply adapted to ONT. We recommend using swabs and DNA extractions protocols with extended washing steps. Both 16S rRNA and metagenomic sequencing achieved reliable and reproducible results. Our benchmarking experiments reveal thresholds for analysis parameters that achieved excellent precision, recall, and area under the precision recall values and is superior to existing classifiers (Kraken2, Kaiju, and MetaMaps). Hence, our workflow provides an experimental and bioinformatic pipeline to perform a highly accurate analysis of complex microbial structures from buccal and rectal swabs. IMPORTANCE Advanced microbiome analysis relies on sequencing of short DNA fragments from microorganisms like bacteria, fungi, and viruses. More recently, long fragment DNA sequencing of 3rd generation sequencing has gained increasing importance and can be rapidly conducted within a few hours due to its potential real-time sequencing. However, the analysis and correct identification of the microbiome relies on a multitude of factors, such as the method of sampling, DNA extraction, sequencing, and bioinformatic analysis. Scientists have used different protocols in the past that do not allow us to compare results across different studies and research fields. Here, we provide a comprehensive workflow from DNA extraction, sequencing, and bioinformatic workflow that allows rapid and accurate analysis of human buccal and rectal swabs with reproducible protocols. This workflow can be readily applied by many scientists from various research fields that aim to use long-fragment microbiome sequencing.
Keywords: 16S rRNA; DNA extraction; Kaiju; Kraken2; MetaMaps; Metapont; ONT; Oxford Nanopore Technologies; bioinformatic workflow; buccal swab; eNAT; eSwab; metagenomics; microbiome; rectal swab; sampling; sequencing; storage.
Figures
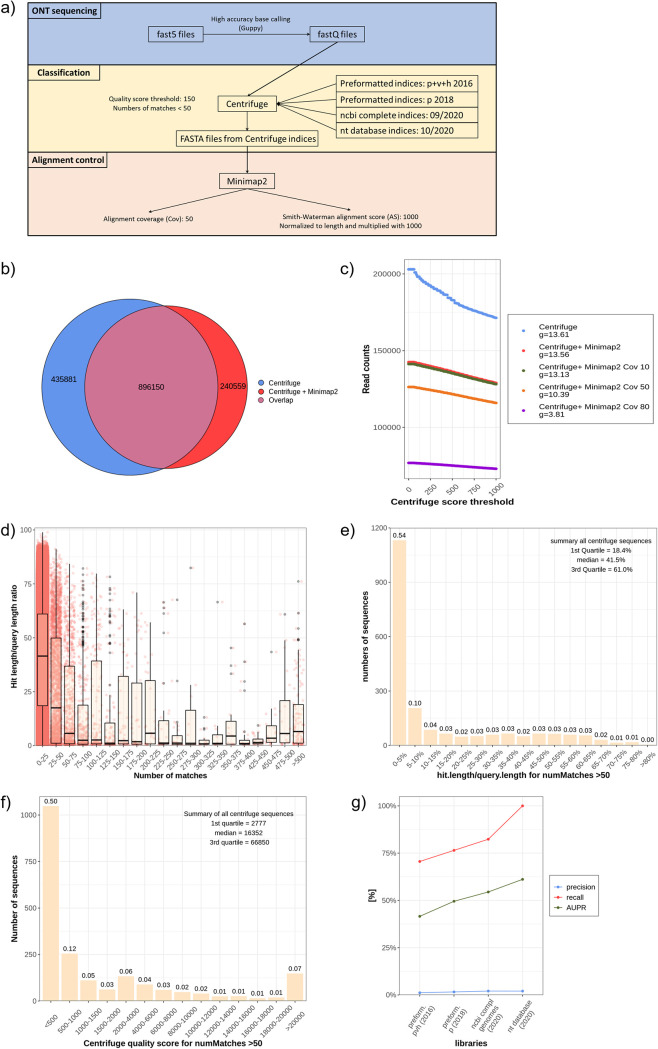
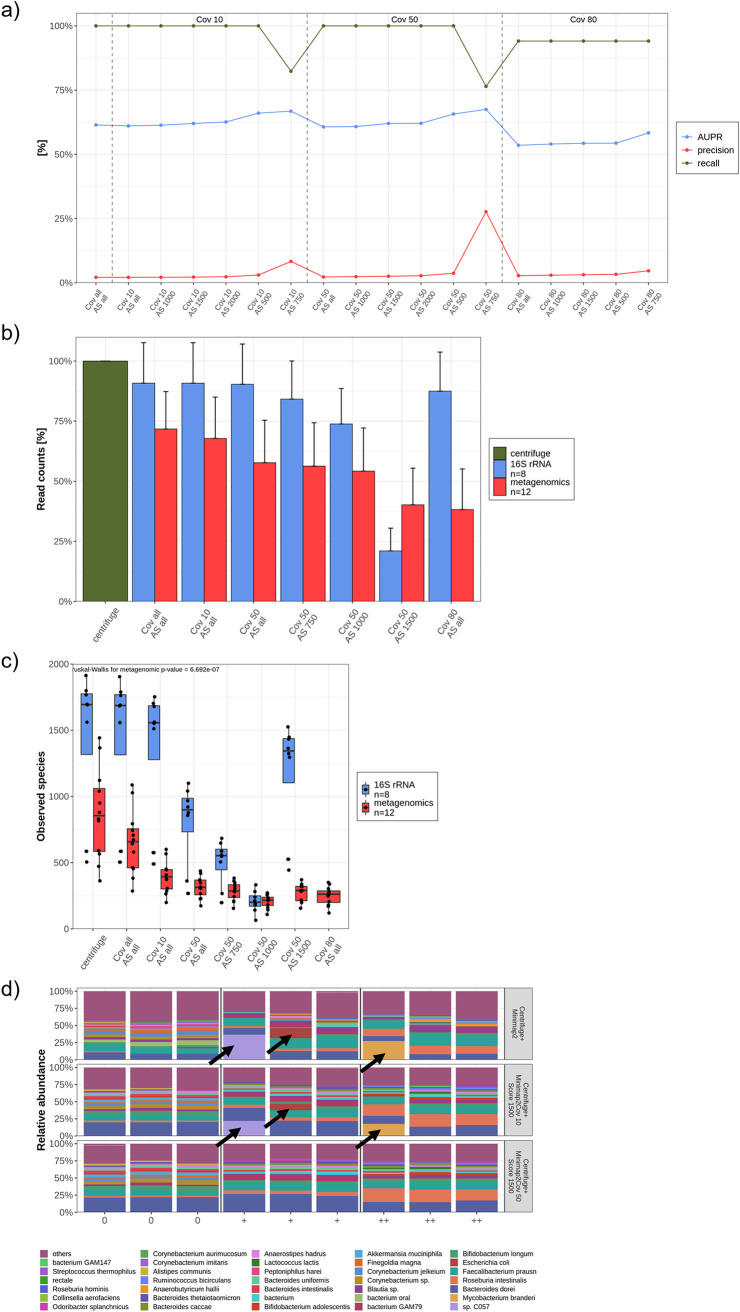
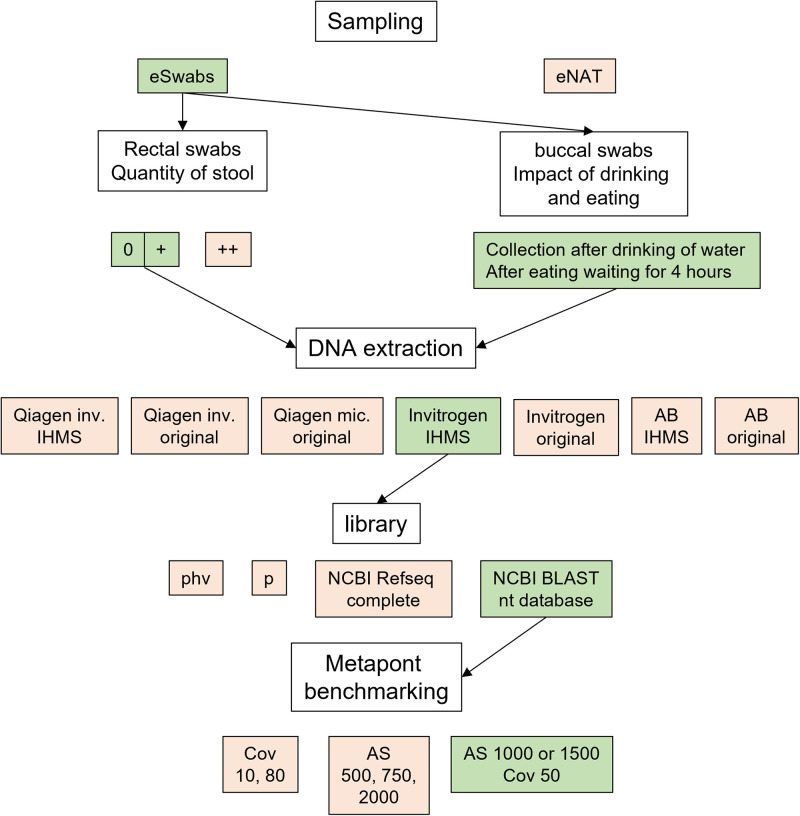
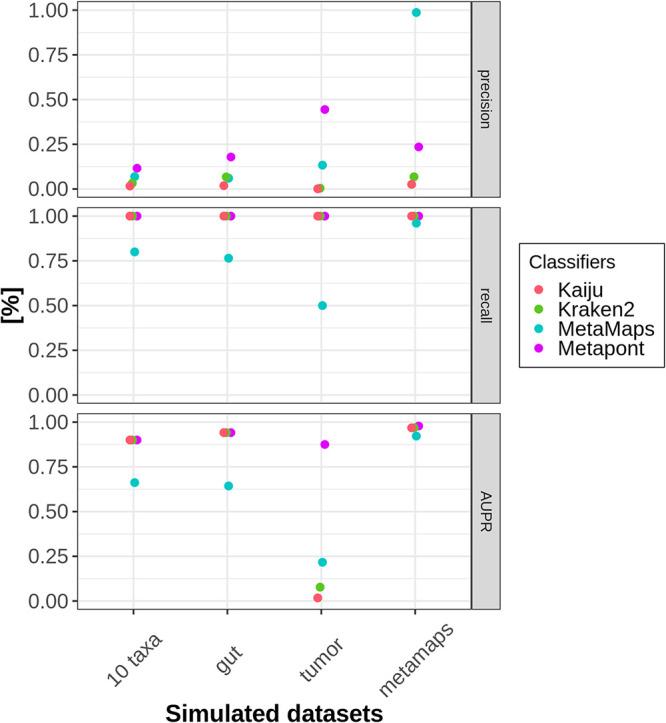
References
-
- Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM, Purdue MP, Abnet CC, Stolzenberg-Solomon R, Miller G, Ravel J, Hayes RB, Ahn J. 2018. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut 67:120–127. doi:10.1136/gutjnl-2016-312580. - DOI - PMC - PubMed
-
- Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, Ståhlman M, Olsson LM, Serino M, Planas-Fèlix M, Xifra G, Mercader JM, Torrents D, Burcelin R, Ricart W, Perkins R, Fernàndez-Real JM, Bäckhed F. 2017. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med 23:850–858. doi:10.1038/nm.4345. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
