

Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1
- PMID: 34427584
- PMCID: PMC8405186
- DOI: 10.1182/bloodadvances.2021004976
Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1
Abstract
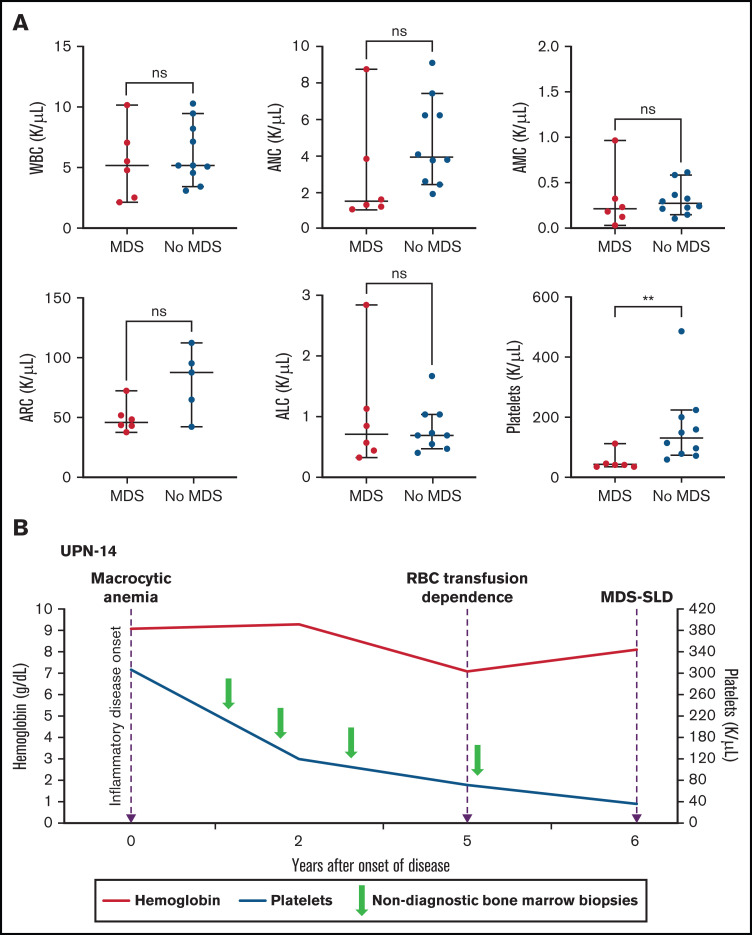
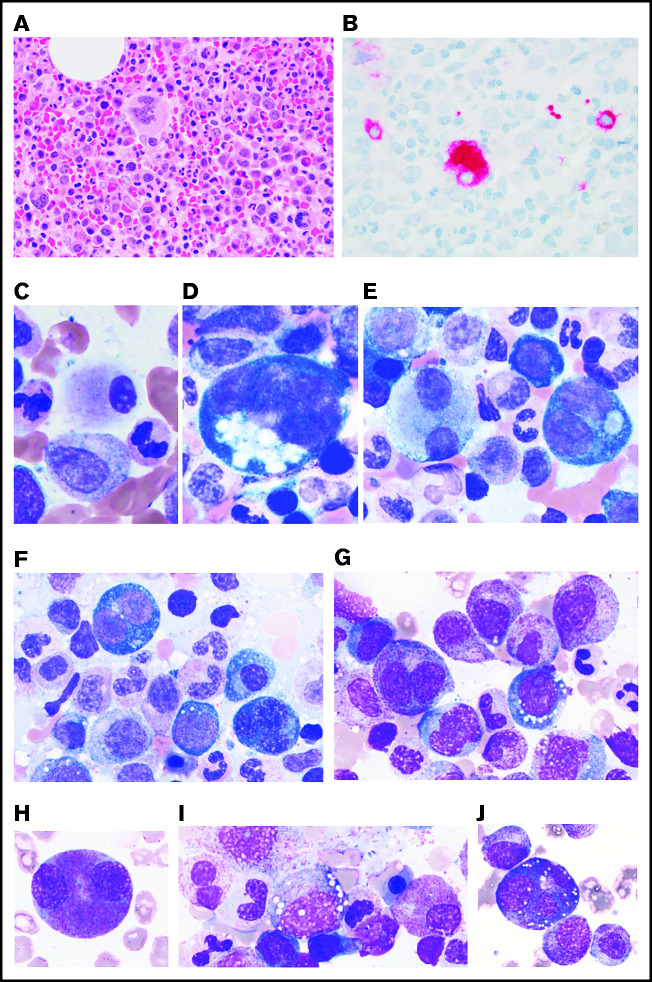
Somatic mutations in UBA1 involving hematopoietic stem and myeloid cells have been reported in patients with the newly defined VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome. Here, we report clinical hematologic manifestations and unique bone marrow (BM) features in 16 patients with VEXAS. All patients were male and had a history of severe autoinflammatory and rheumatologic manifestations and a somatic UBA1 mutation (p.Met41). Ten patients had hematologic disorders: myelodysplastic syndrome (MDS; 6 of 16), multiple myeloma (2 of 16), monoclonal gammopathy of undetermined significance (2 of 16), and monoclonal B-cell lymphocytosis (2 of 16), and a few of those patients had 2 co-existing clonal processes. Although macrocytic anemia (100%) and lymphopenia (80%) were prevalent in all patients with VEXAS, thrombocytopenia and neutropenia were more common in patients with progression to MDS. All BMs in VEXAS patients had prominent cytoplasmic vacuoles in myeloid and erythroid precursors. In addition, most BMs were hypercellular with myeloid hyperplasia, erythroid hypoplasia, and varying degrees of dysplasia. All patients diagnosed with MDS were lower risk (low blast count, very good to intermediate cytogenetics) according to standard prognostic scoring with no known progression to leukemia. In addition, 10 of 16 patients had thrombotic events, including venous thromboembolism and arterial stroke. Although VEXAS presents symptomatically as a rheumatologic disease, morbidity and mortality are associated with progression to hematologic disease. Given the increased risk of developing MDS and multiple myeloma, surveillance for disease progression is important.
© 2021 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures



References
-
- Enright H, Jacob HS, Vercellotti G, Howe R, Belzer M, Miller W.. Paraneoplastic autoimmune phenomena in patients with myelodysplastic syndromes: response to immunosuppressive therapy. Br J Haematol. 1995;91(2):403-408. - PubMed
-
- Billström R, Johansson H, Johansson B, Mitelman F.. Immune-mediated complications in patients with myelodysplastic syndromes--clinical and cytogenetic features. Eur J Haematol. 1995;55(1):42-48. - PubMed
-
- Fain O, Hamidou M, Cacoub P, et al. . Vasculitides associated with malignancies: analysis of sixty patients. Arthritis Rheum. 2007;57(8):1473-1480. - PubMed
-
- de Hollanda A, Beucher A, Henrion D, et al. . Systemic and immune manifestations in myelodysplasia: a multicenter retrospective study. Arthritis Care Res (Hoboken). 2011;63(8):1188-1194. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
