

TERA-Seq: true end-to-end sequencing of native RNA molecules for transcriptome characterization
- PMID: 34428294
- PMCID: PMC8599856
- DOI: 10.1093/nar/gkab713
TERA-Seq: true end-to-end sequencing of native RNA molecules for transcriptome characterization
Abstract
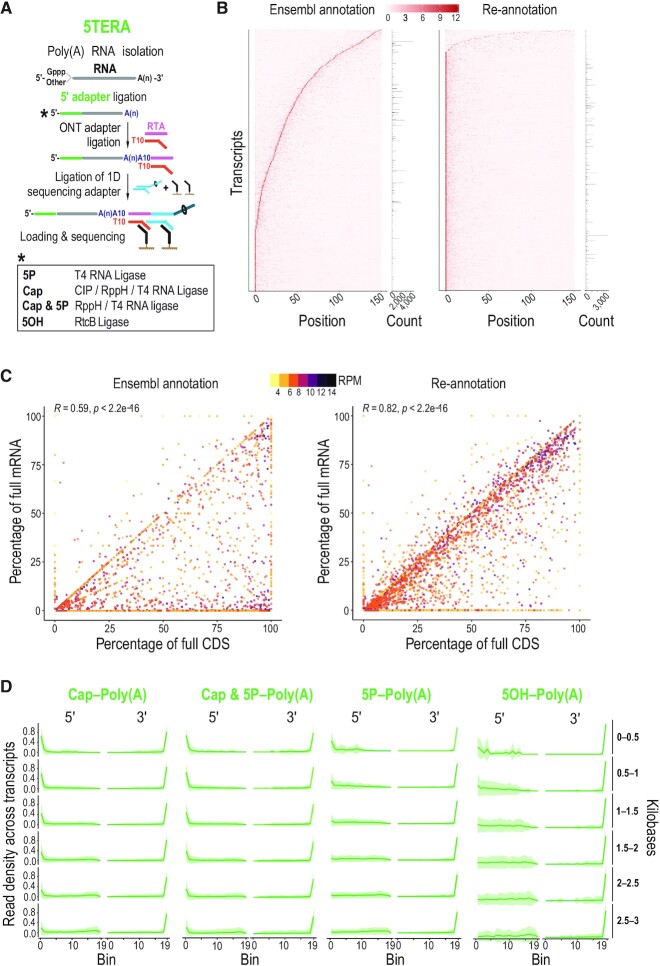
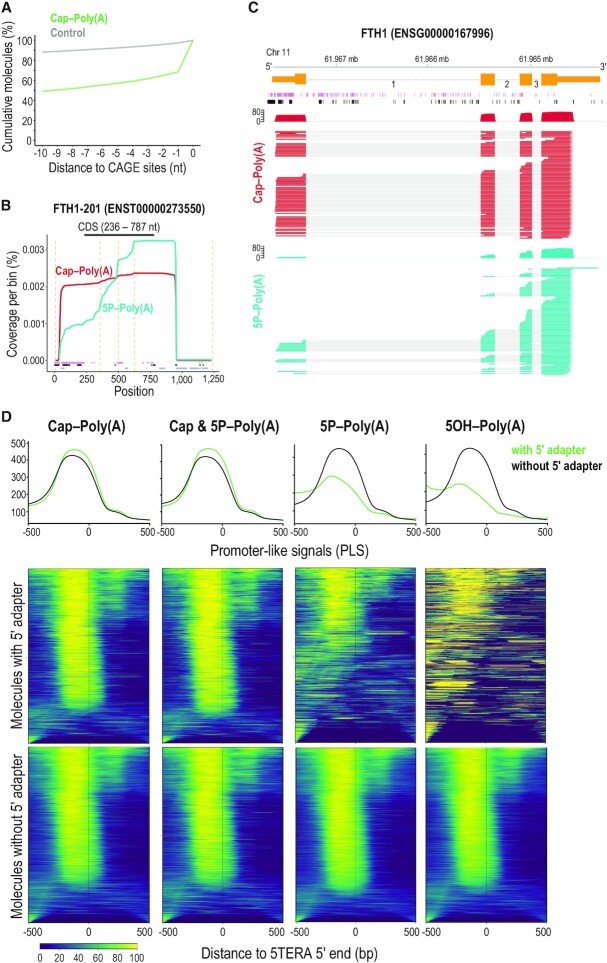
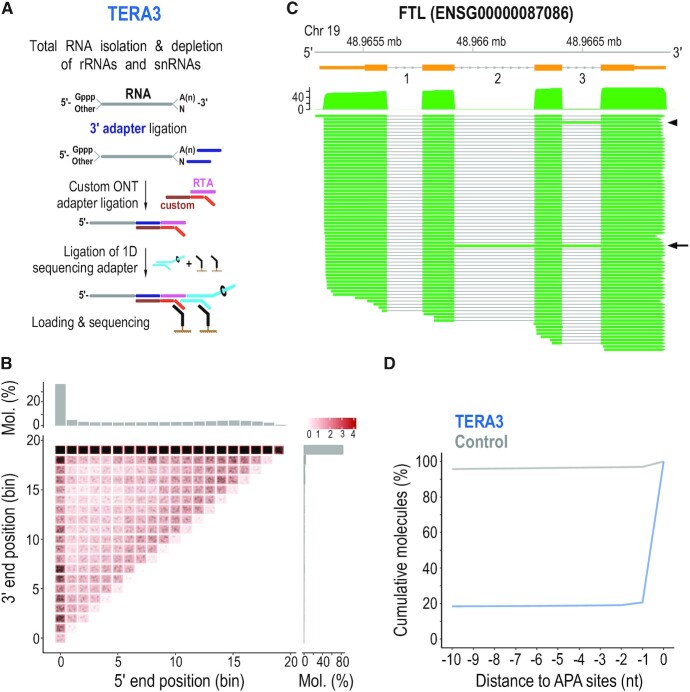
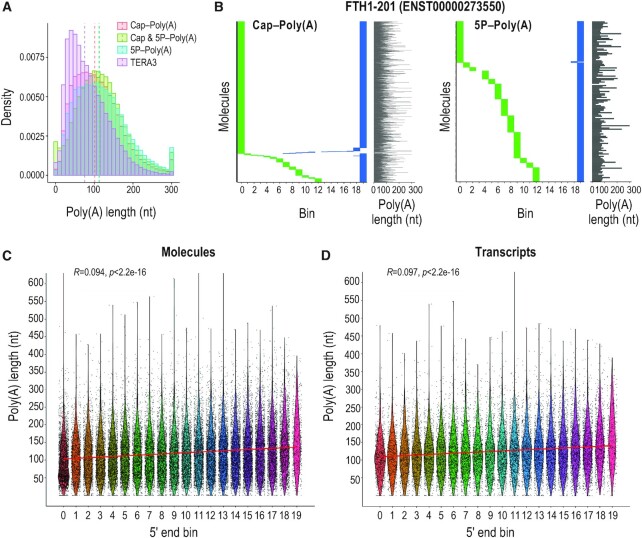
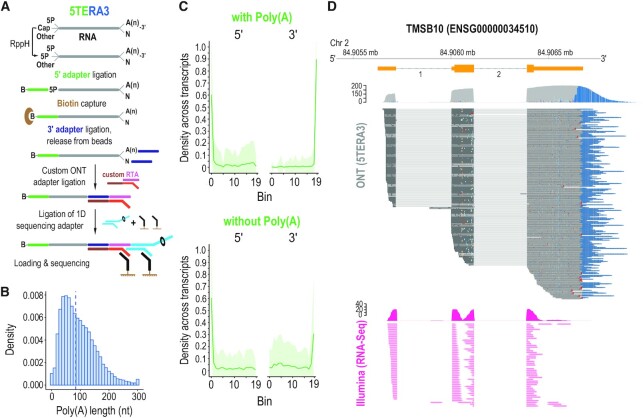
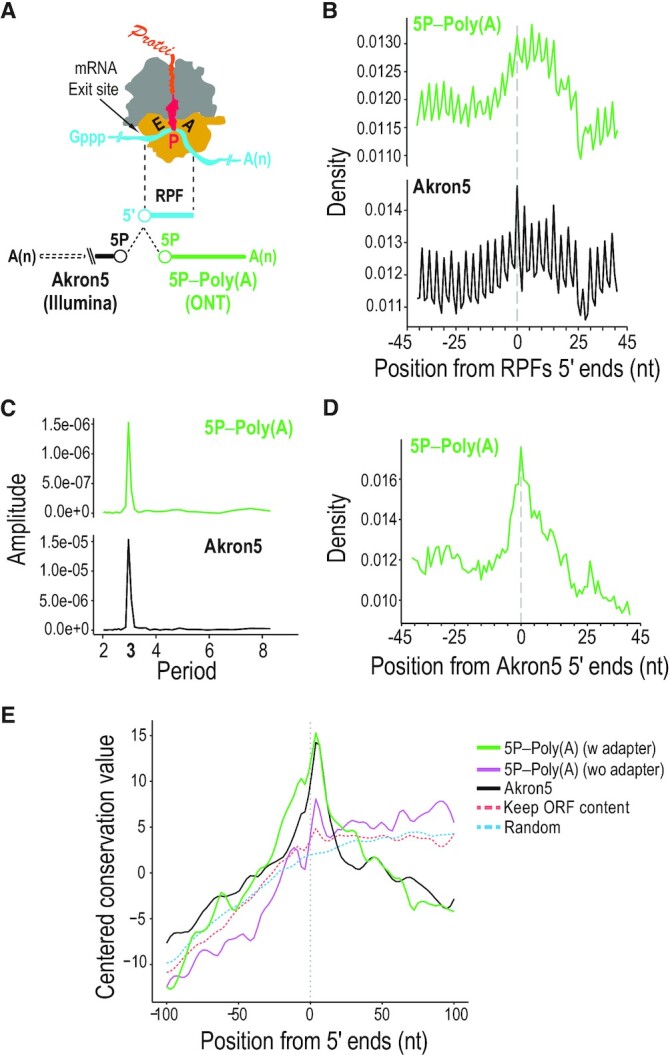
Direct sequencing of single, native RNA molecules through nanopores has a strong potential to transform research in all aspects of RNA biology and clinical diagnostics. The existing platform from Oxford Nanopore Technologies is unable to sequence the very 5' ends of RNAs and is limited to polyadenylated molecules. Here, we develop True End-to-end RNA Sequencing (TERA-Seq), a platform that addresses these limitations, permitting more thorough transcriptome characterization. TERA-Seq describes both poly- and non-polyadenylated RNA molecules and accurately identifies their native 5' and 3' ends by ligating uniquely designed adapters that are sequenced along with the transcript. We find that capped, full-length mRNAs in human cells show marked variation of poly(A) tail lengths at the single molecule level. We report prevalent capping downstream of canonical transcriptional start sites in otherwise fully spliced and polyadenylated molecules. We reveal RNA processing and decay at single molecule level and find that mRNAs decay cotranslationally, often from their 5' ends, while frequently retaining poly(A) tails. TERA-Seq will prove useful in many applications where true end-to-end direct sequencing of single, native RNA molecules and their isoforms is desirable.
© The Author(s) 2021. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Krebs J.E., Goldstein E.S., Kilpatrick S.T.. Lewin's Genes. 2018; 12th edn.Jones & Bartlett Learning.
-
- Isken O., Maquat L.E.. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007; 21:1833–1856. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
