

A clinically applicable integrative molecular classification of meningiomas
- PMID: 34433969
- PMCID: PMC11604310
- DOI: 10.1038/s41586-021-03850-3
A clinically applicable integrative molecular classification of meningiomas
Abstract
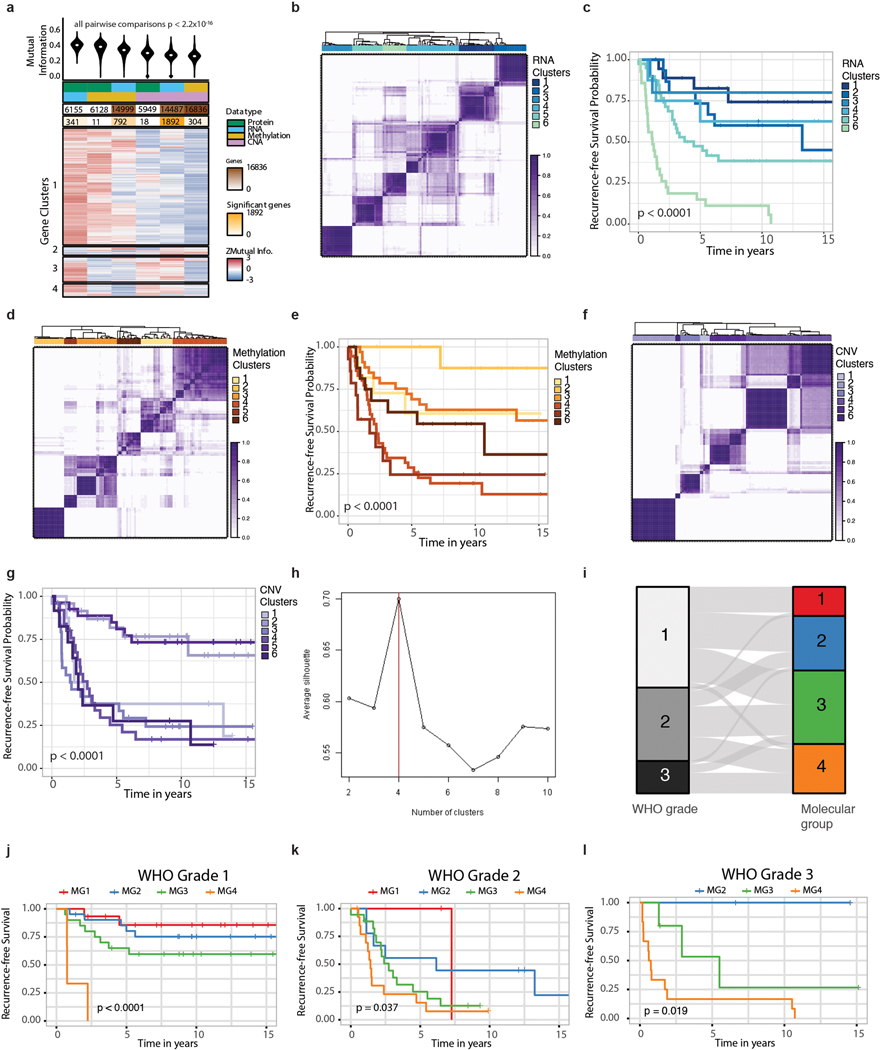
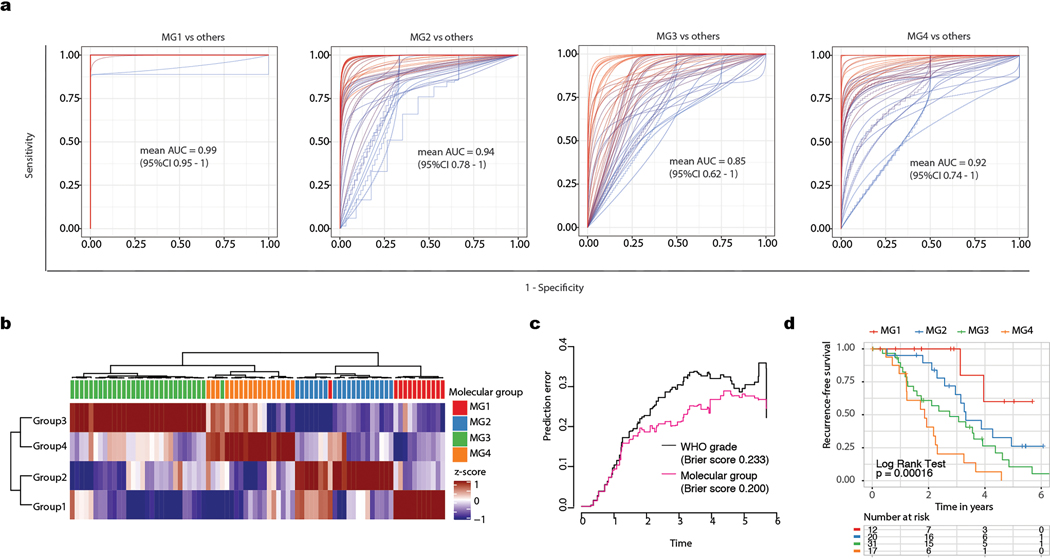
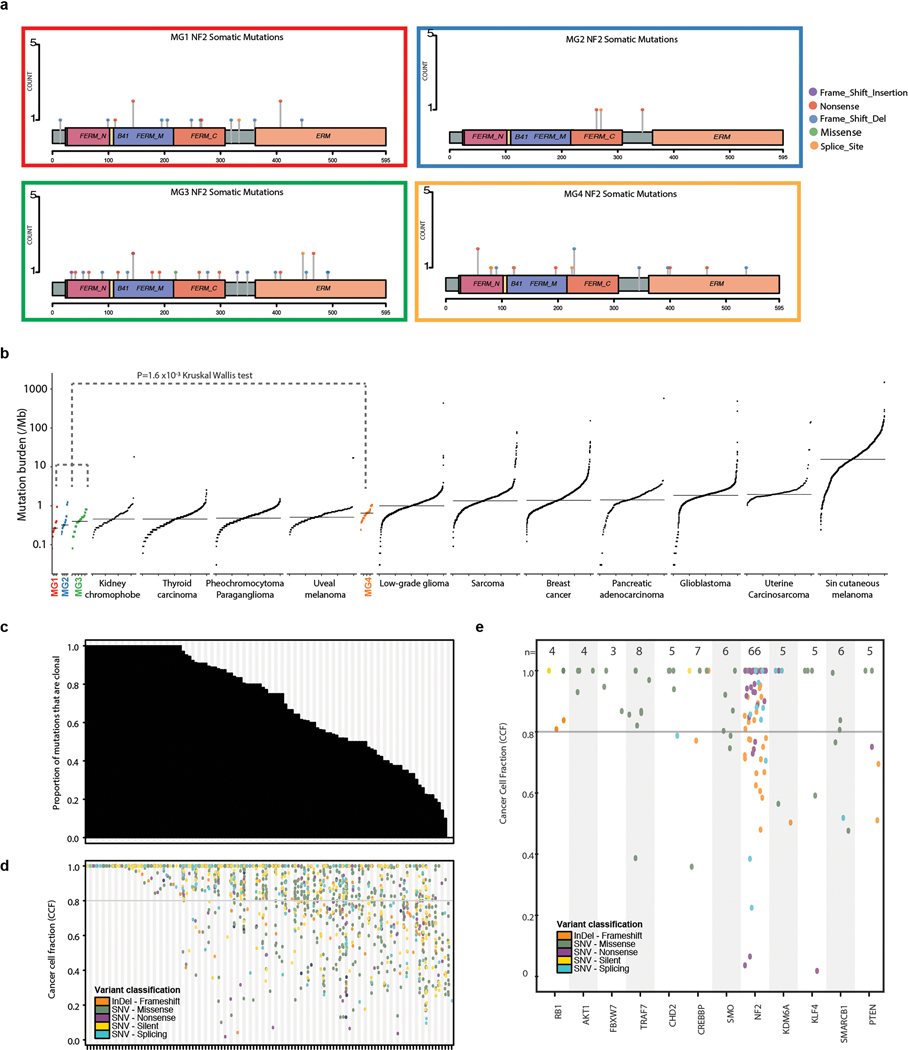
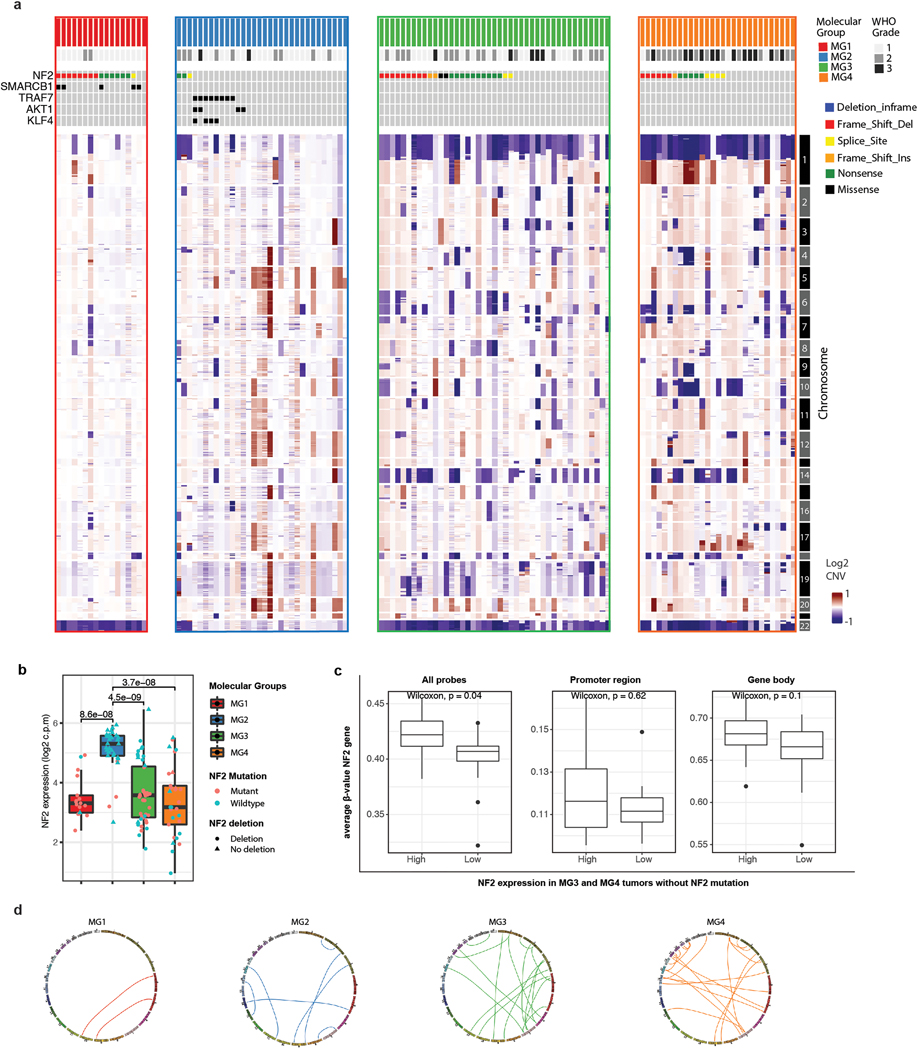
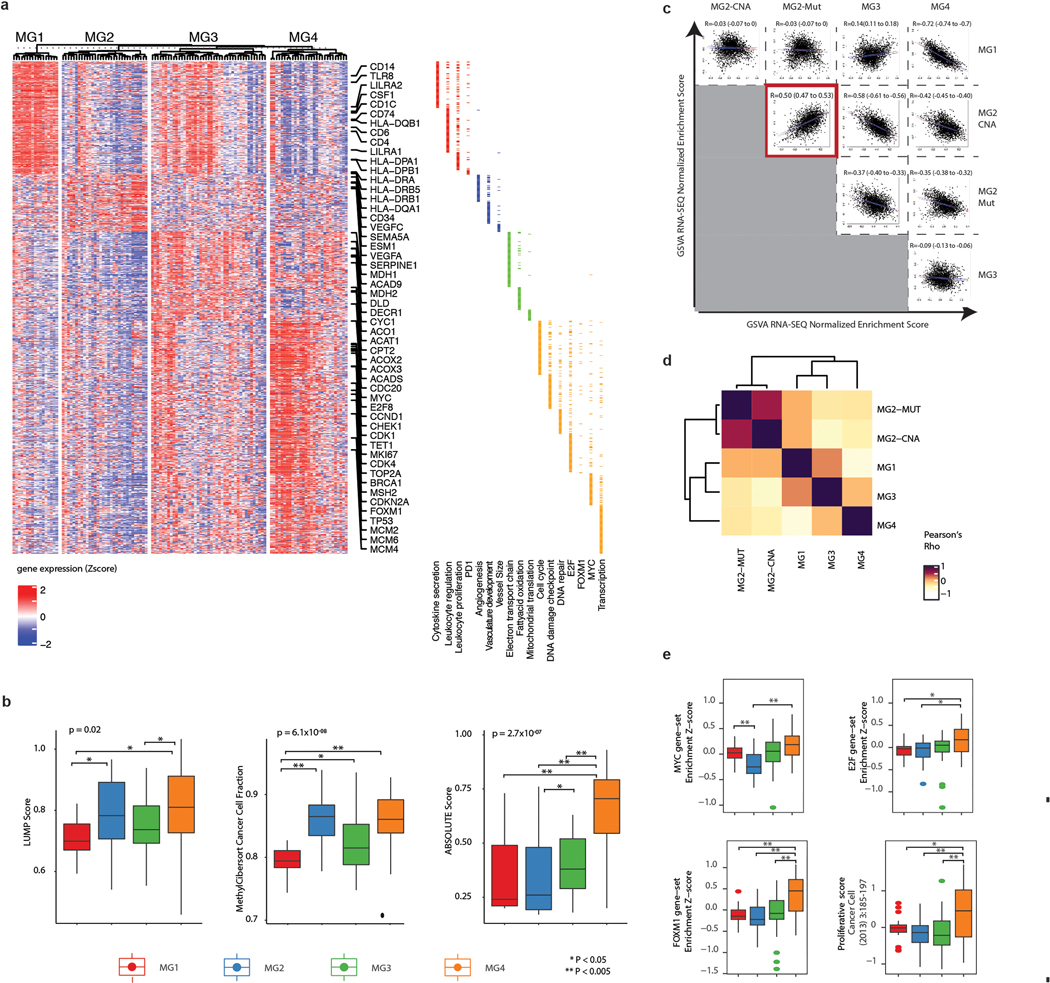
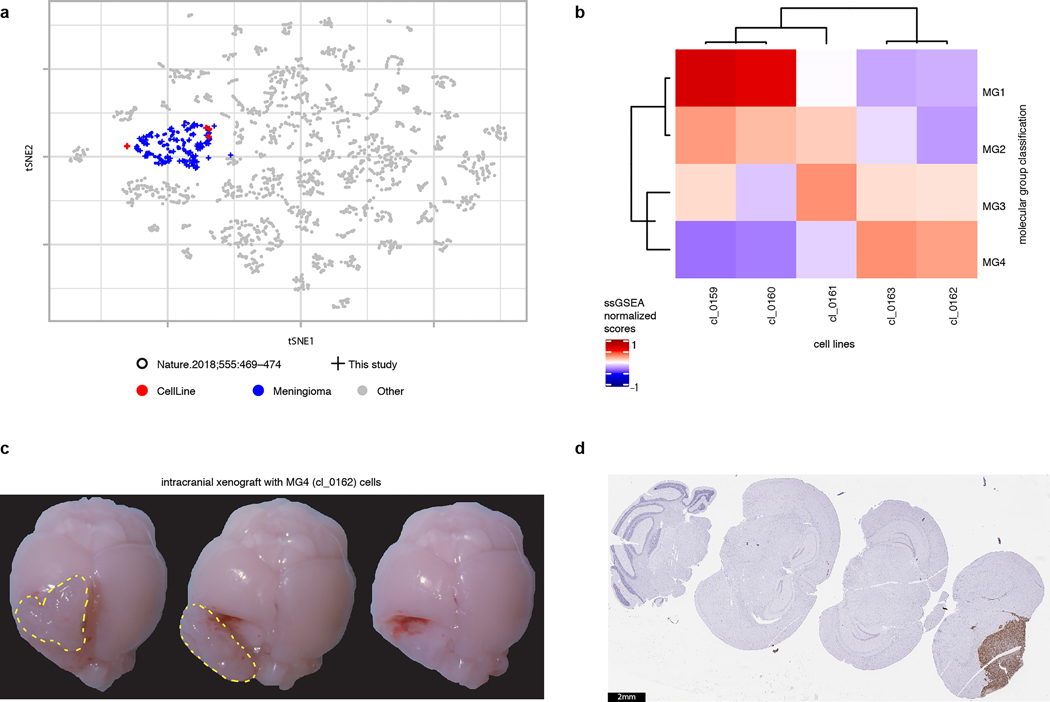
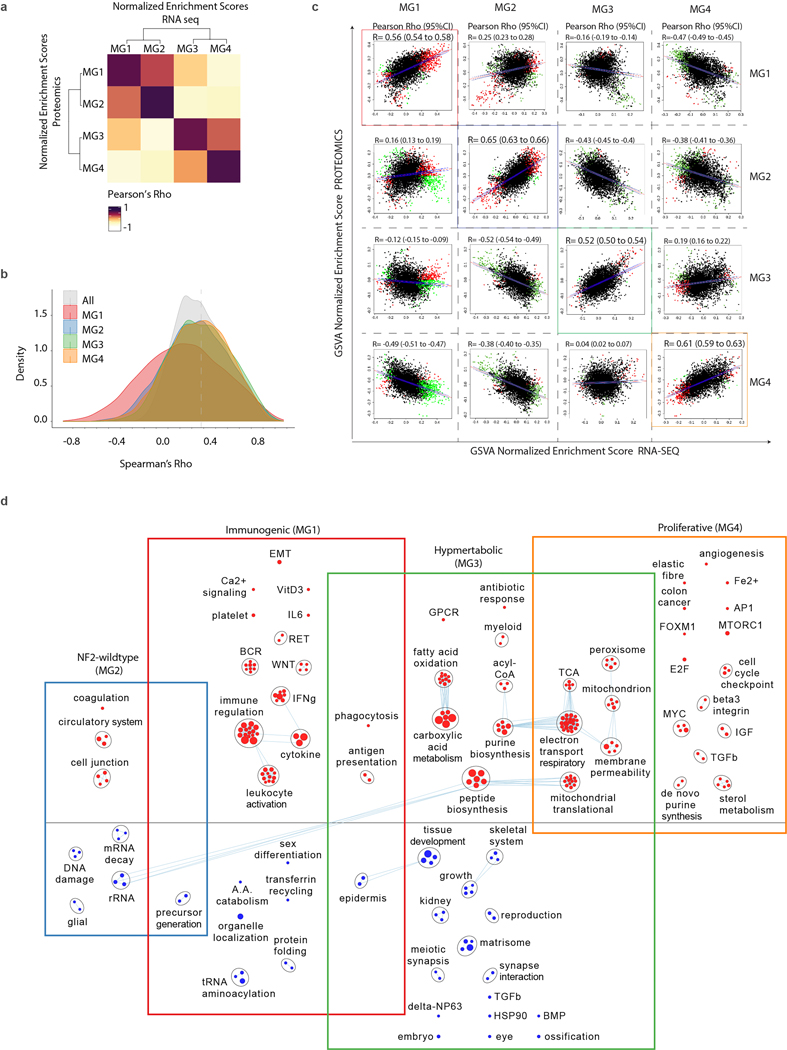
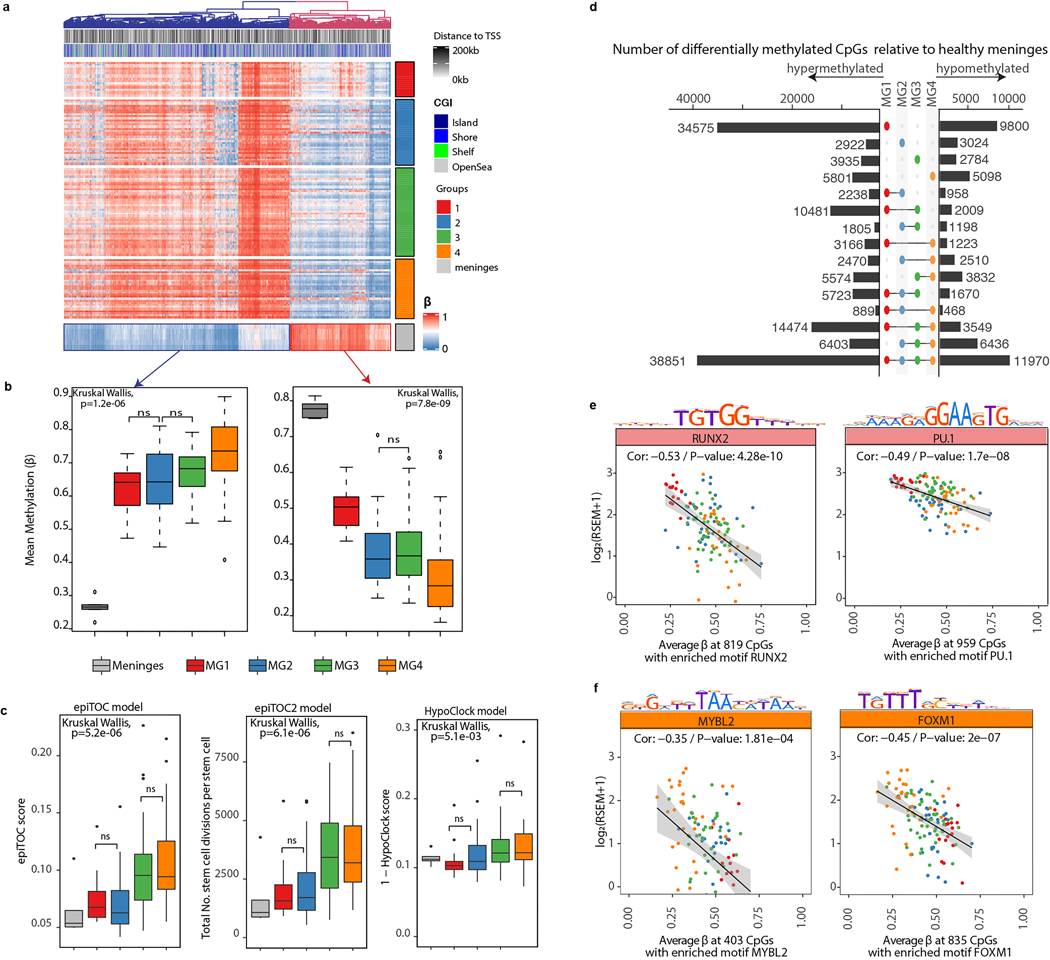
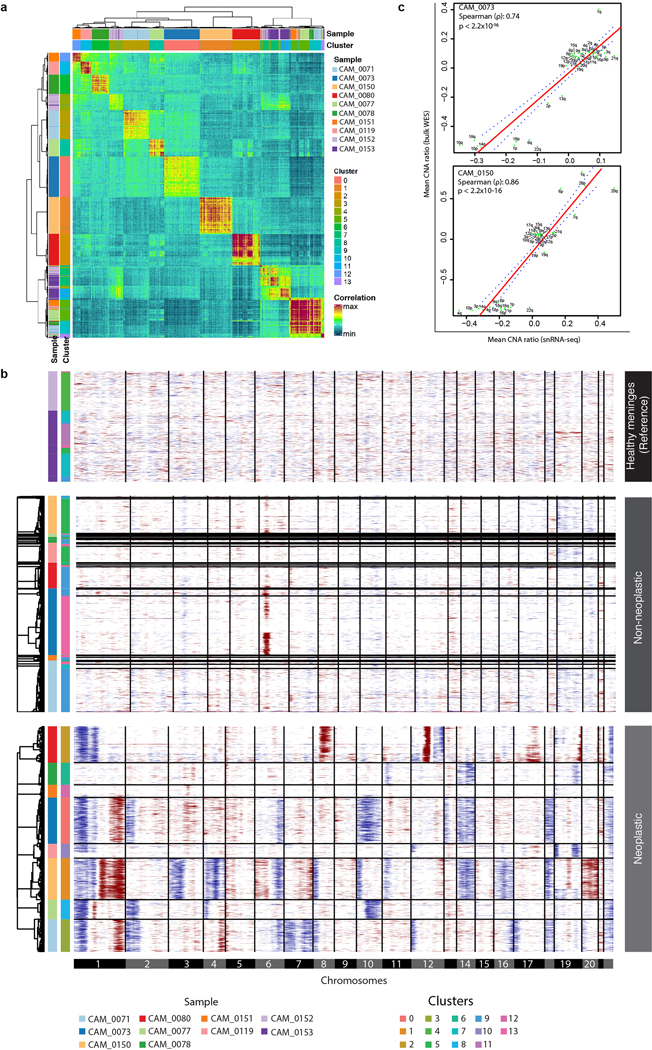
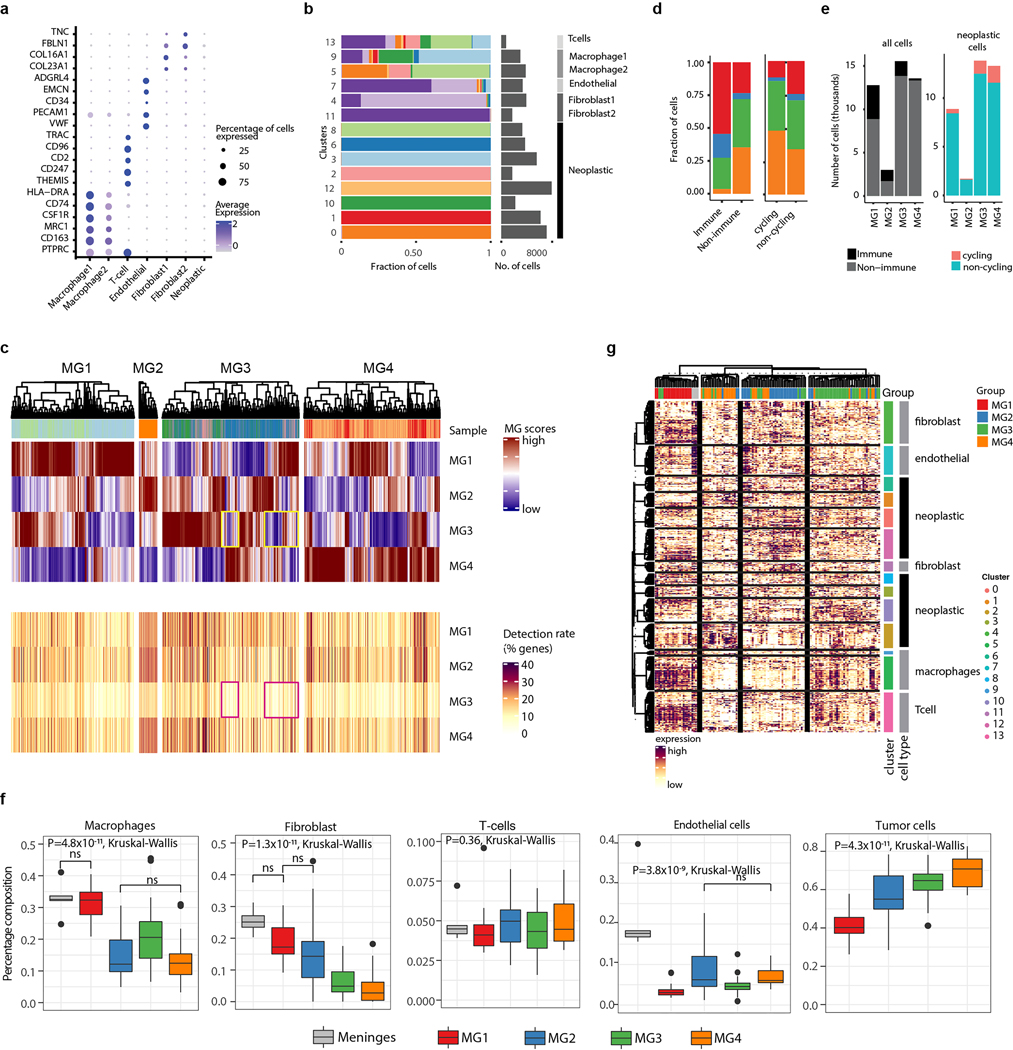
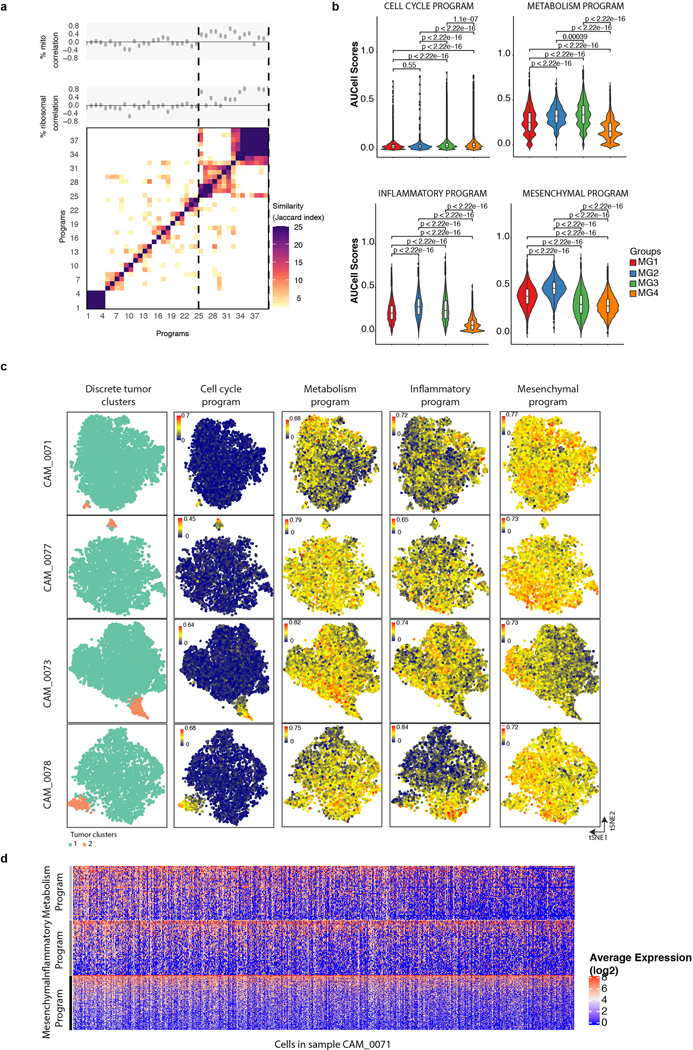
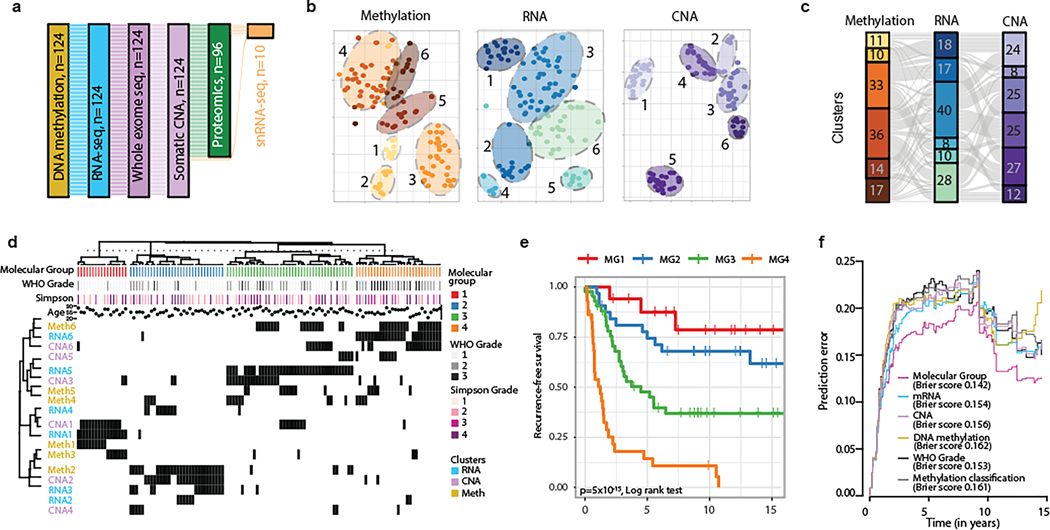
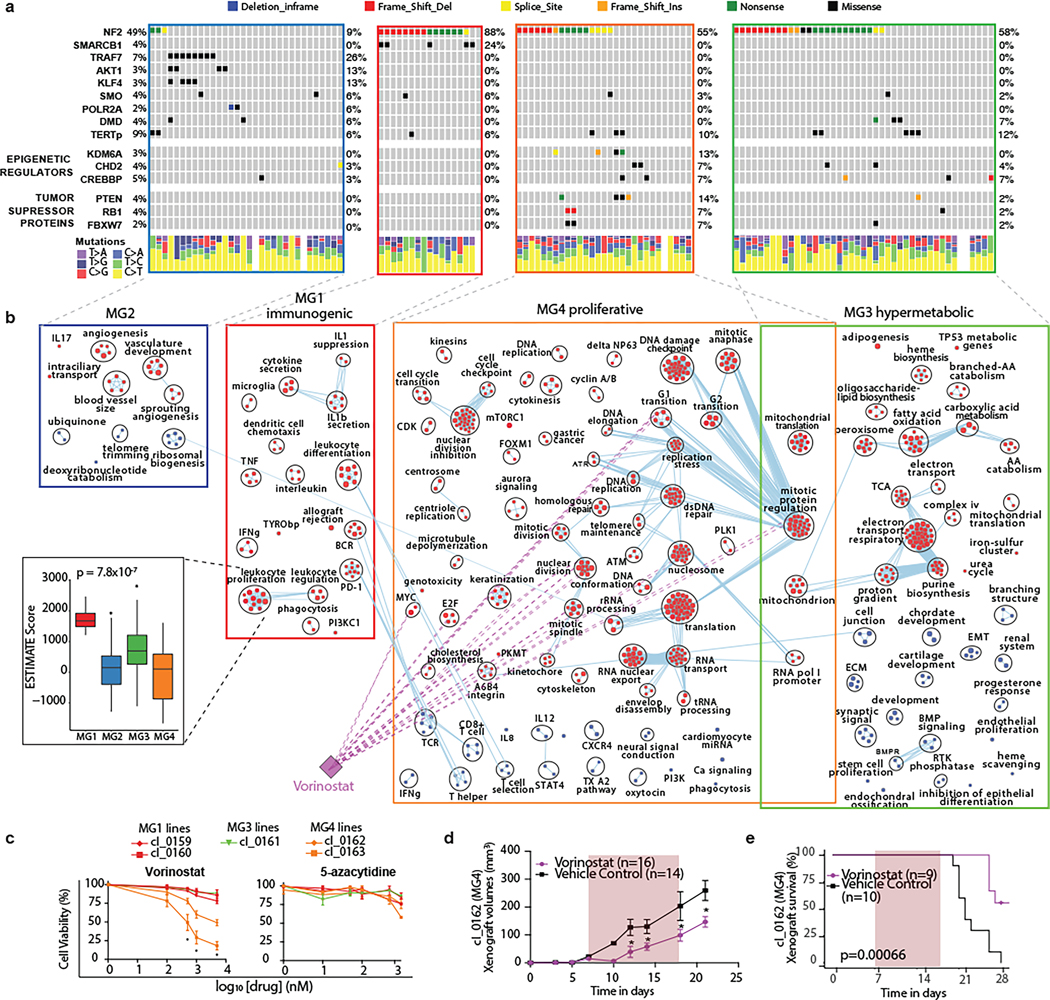
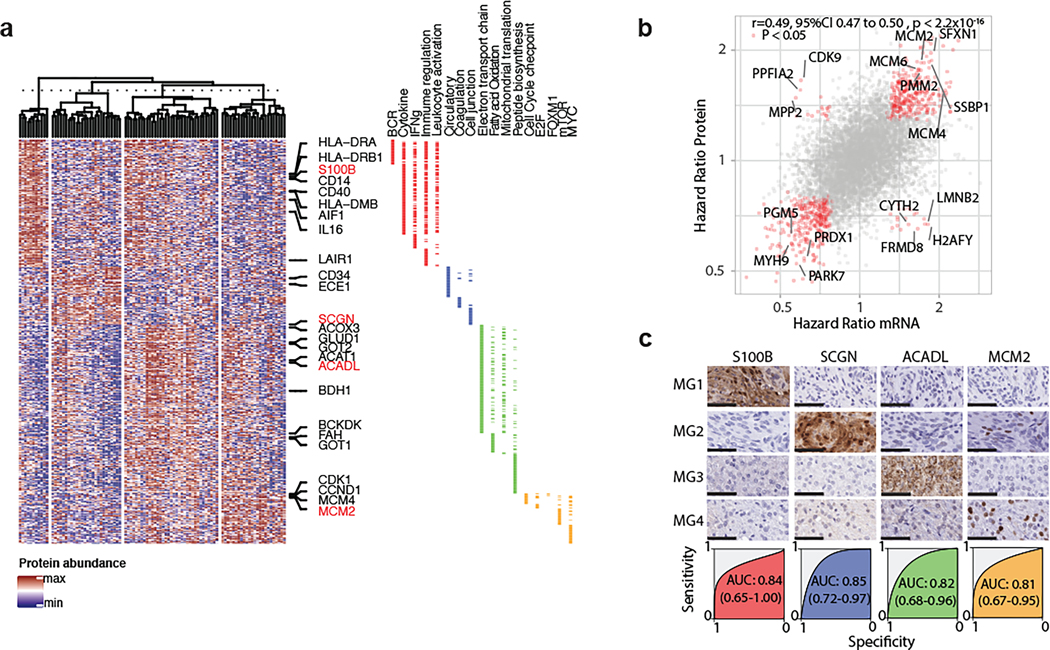
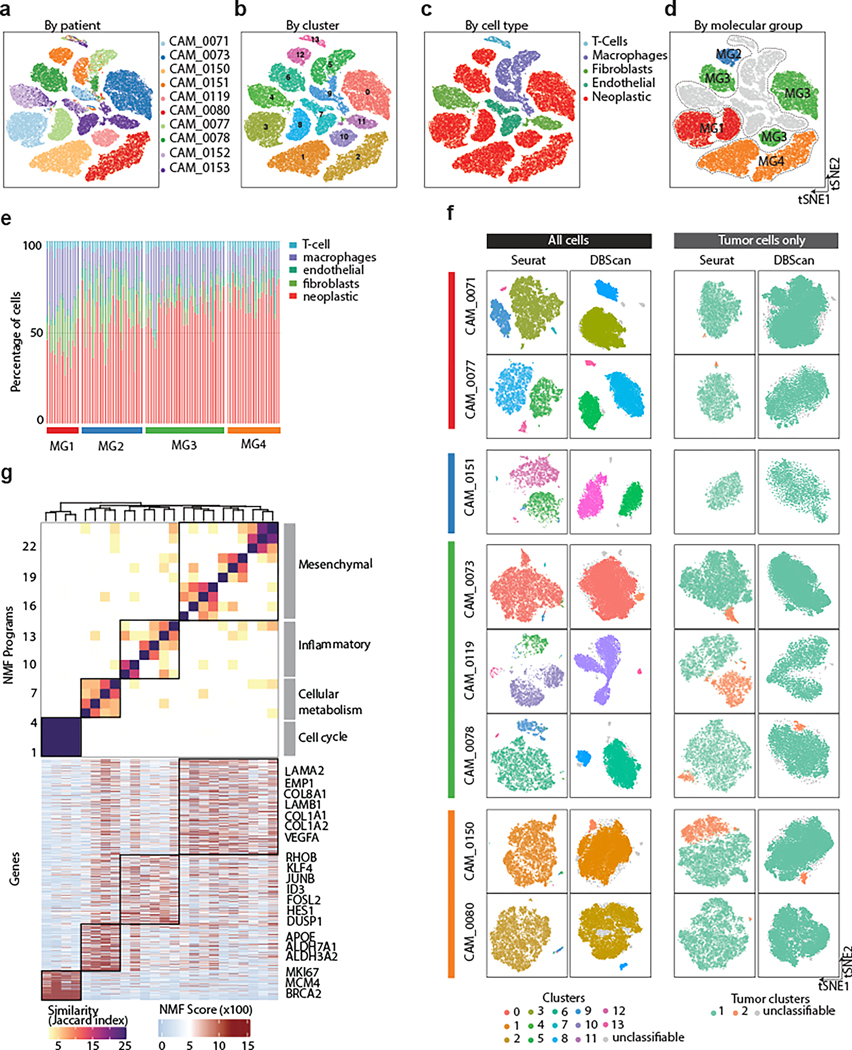
Meningiomas are the most common primary intracranial tumour in adults1. Patients with symptoms are generally treated with surgery as there are no effective medical therapies. The World Health Organization histopathological grade of the tumour and the extent of resection at surgery (Simpson grade) are associated with the recurrence of disease; however, they do not accurately reflect the clinical behaviour of all meningiomas2. Molecular classifications of meningioma that reliably reflect tumour behaviour and inform on therapies are required. Here we introduce four consensus molecular groups of meningioma by combining DNA somatic copy-number aberrations, DNA somatic point mutations, DNA methylation and messenger RNA abundance in a unified analysis. These molecular groups more accurately predicted clinical outcomes compared with existing classification schemes. Each molecular group showed distinctive and prototypical biology (immunogenic, benign NF2 wild-type, hypermetabolic and proliferative) that informed therapeutic options. Proteogenomic characterization reinforced the robustness of the newly defined molecular groups and uncovered highly abundant and group-specific protein targets that we validated using immunohistochemistry. Single-cell RNA sequencing revealed inter-individual variations in meningioma as well as variations in intrinsic expression programs in neoplastic cells that mirrored the biology of the molecular groups identified.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
D.D.D.C., and A.C. are listed as inventors on patents filed that are unrelated to this project. D.D.D.C. received research funding from Pfizer and Nektar therapeutics not related to this project.
Figures

References
References (Methods only)
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
