

The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children
- PMID: 34437303
- PMCID: PMC8516454
- DOI: 10.1172/JCI151520
The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children
Abstract
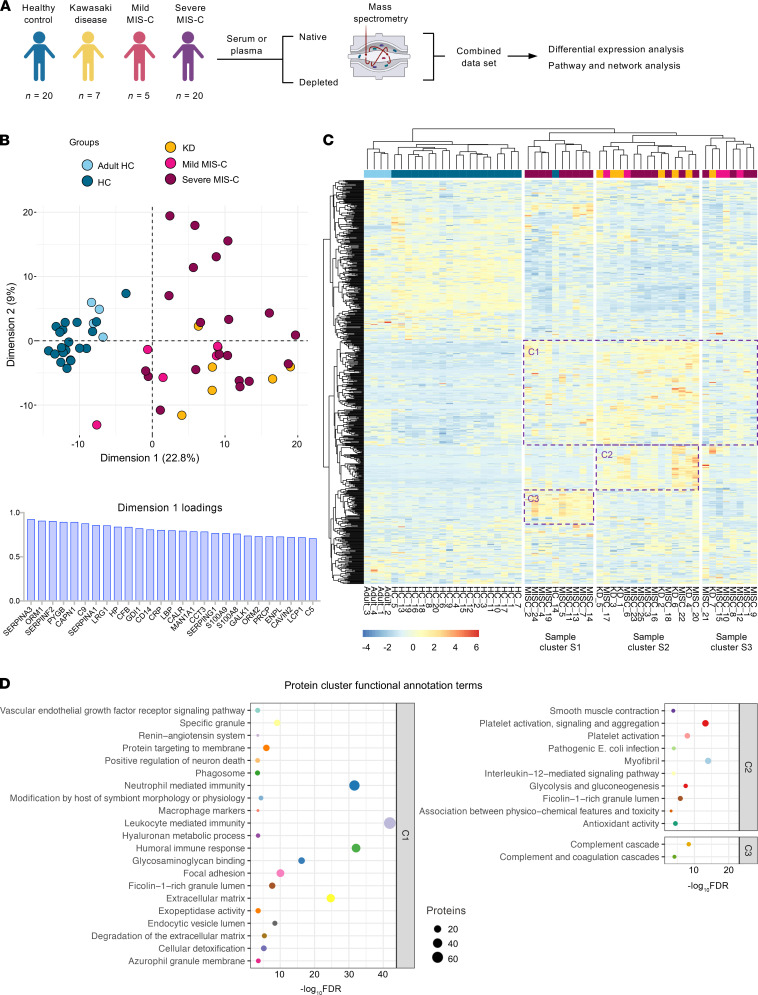
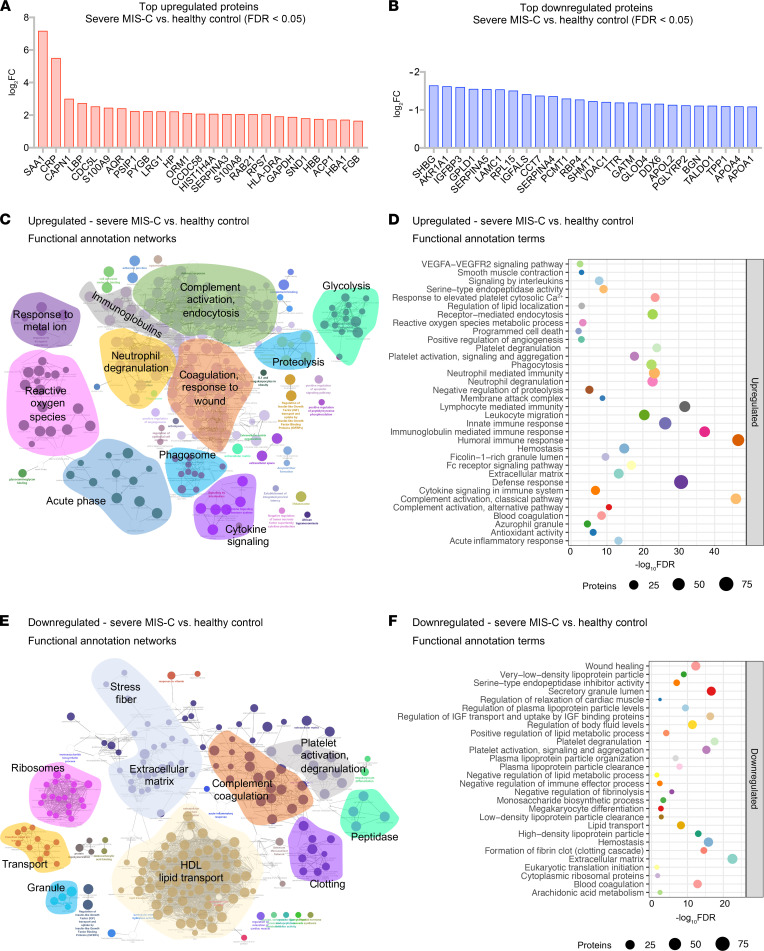
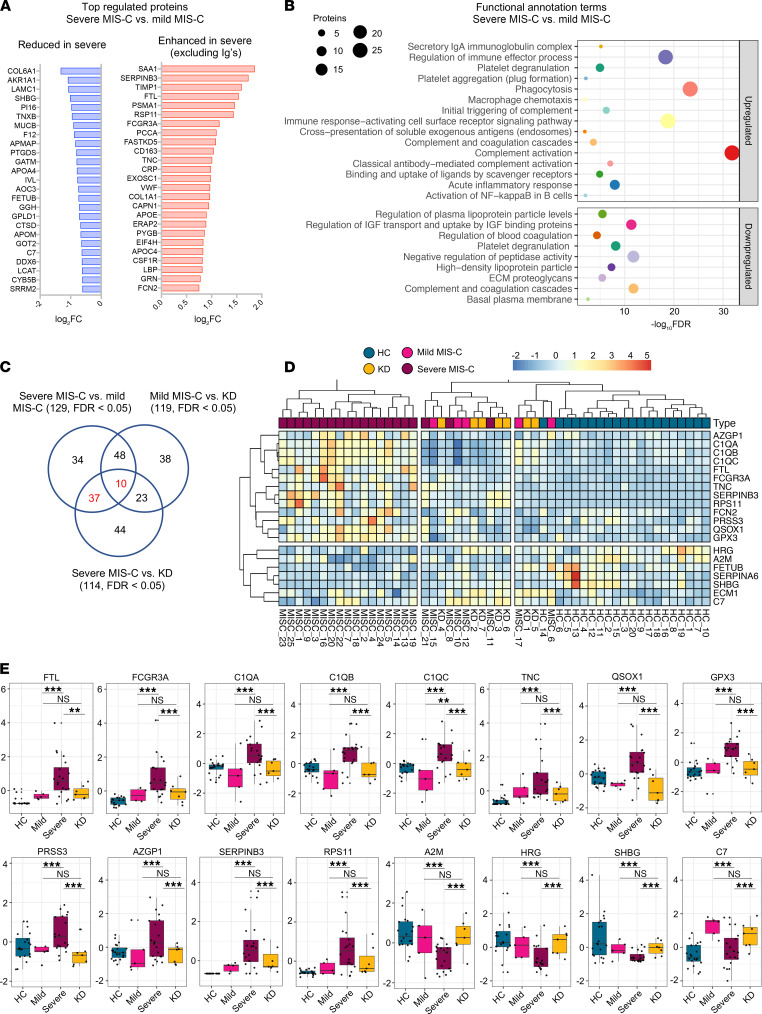
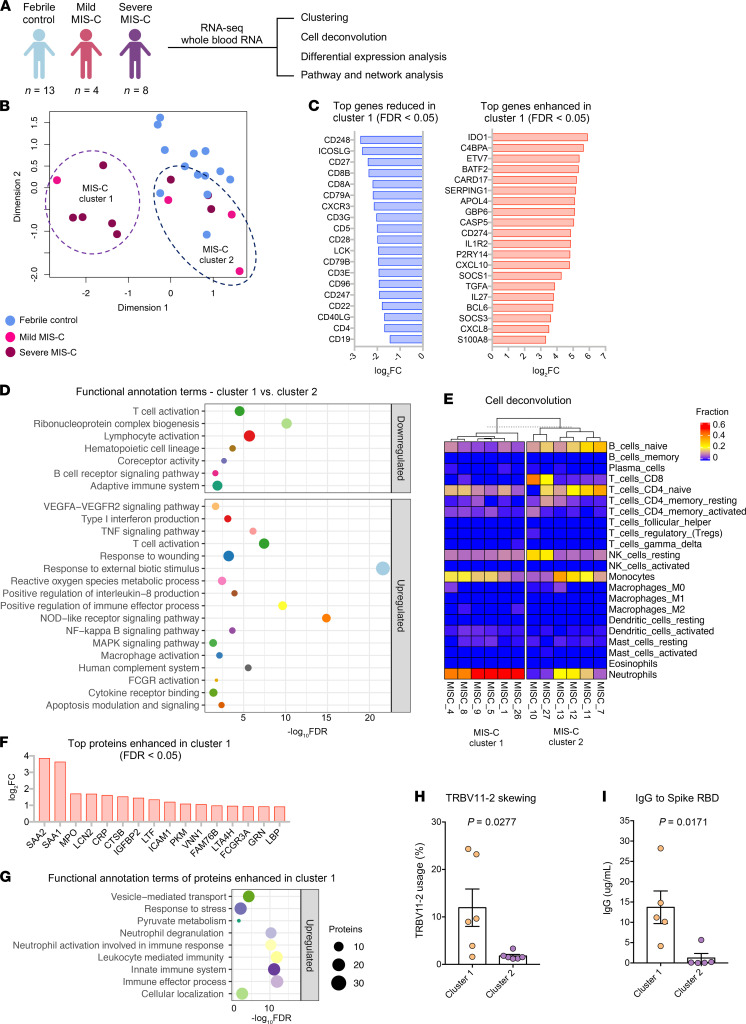
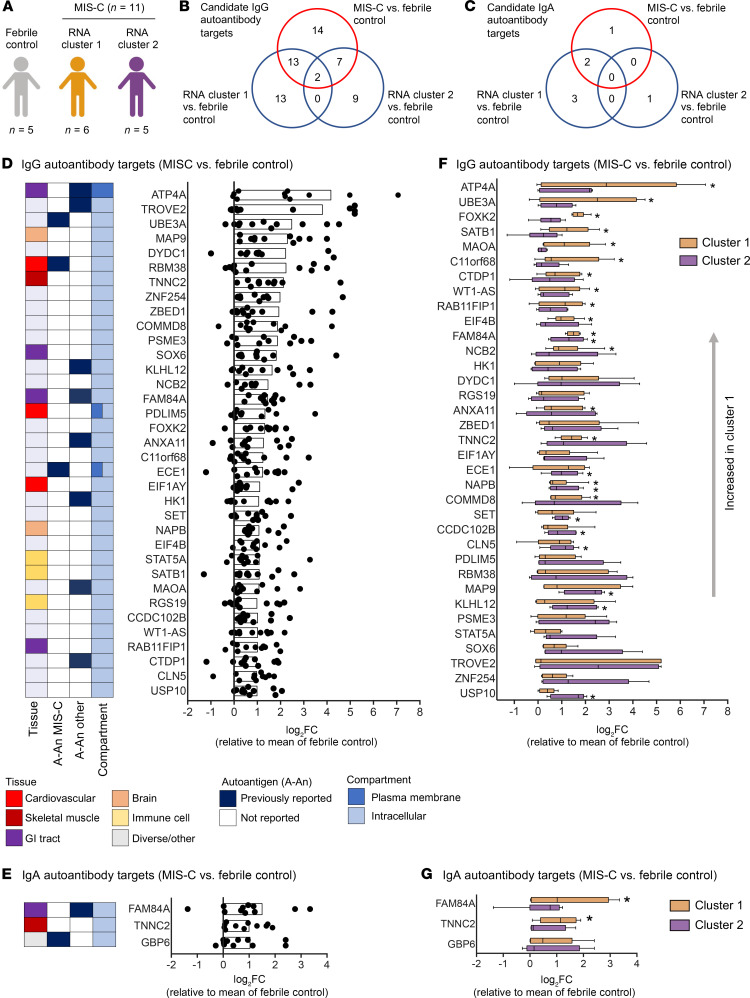
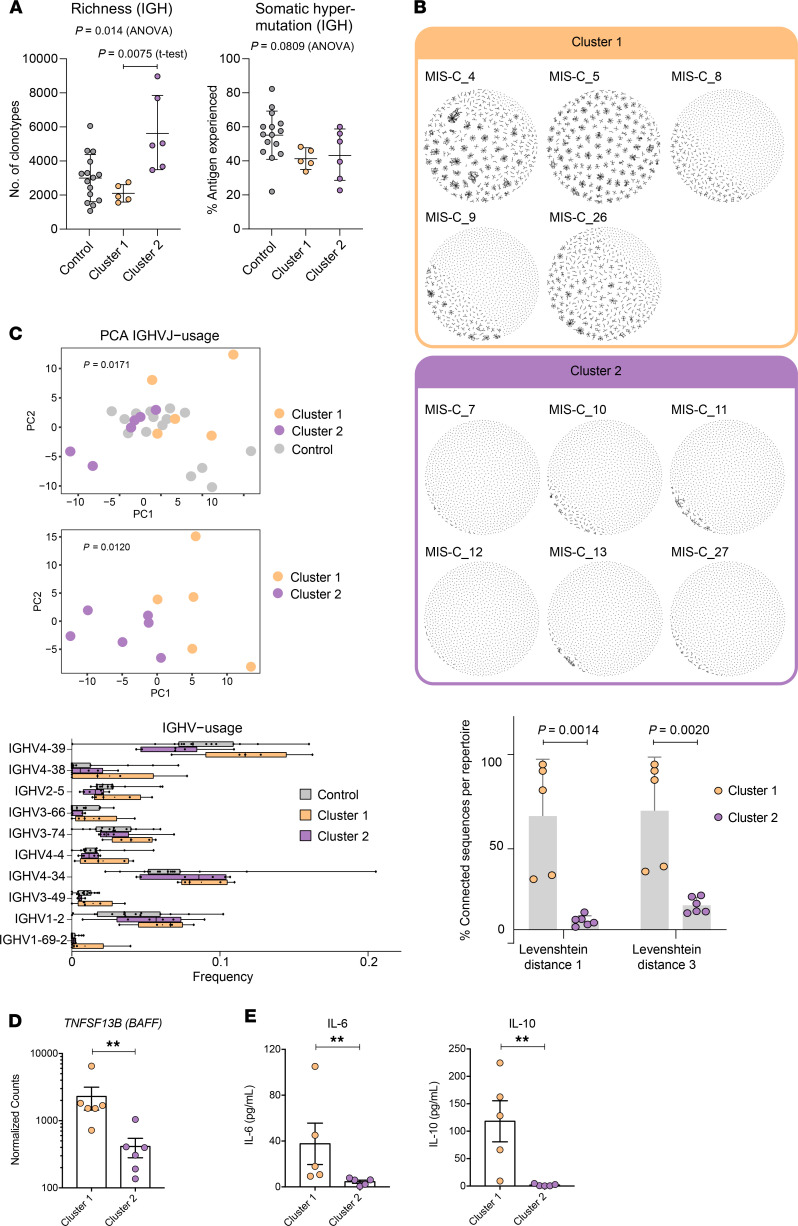
Multisystem inflammatory syndrome in children (MIS-C) manifests as a severe and uncontrolled inflammatory response with multiorgan involvement, occurring weeks after SARS-CoV-2 infection. Here, we utilized proteomics, RNA sequencing, autoantibody arrays, and B cell receptor (BCR) repertoire analysis to characterize MIS-C immunopathogenesis and identify factors contributing to severe manifestations and intensive care unit admission. Inflammation markers, humoral immune responses, neutrophil activation, and complement and coagulation pathways were highly enriched in MIS-C patient serum, with a more hyperinflammatory profile in severe than in mild MIS-C cases. We identified a strong autoimmune signature in MIS-C, with autoantibodies targeted to both ubiquitously expressed and tissue-specific antigens, suggesting autoantigen release and excessive antigenic drive may result from systemic tissue damage. We further identified a cluster of patients with enhanced neutrophil responses as well as high anti-Spike IgG and autoantibody titers. BCR sequencing of these patients identified a strong imprint of antigenic drive with substantial BCR sequence connectivity and usage of autoimmunity-associated immunoglobulin heavy chain variable region (IGHV) genes. This cluster was linked to a TRBV11-2 expanded T cell receptor (TCR) repertoire, consistent with previous studies indicating a superantigen-driven pathogenic process. Overall, we identify a combination of pathogenic pathways that culminate in MIS-C and may inform treatment.
Keywords: Autoimmune diseases; COVID-19; Cellular immune response; Cytokines; Inflammation.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
