

Advances in Genetic and Molecular Understanding of Alzheimer's Disease
- PMID: 34440421
- PMCID: PMC8394321
- DOI: 10.3390/genes12081247
Advances in Genetic and Molecular Understanding of Alzheimer's Disease
Abstract
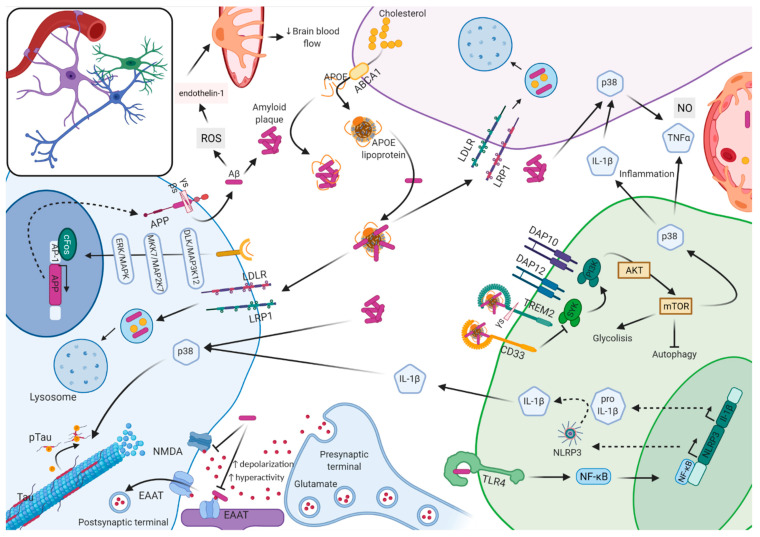
Alzheimer's disease (AD) has become a common disease of the elderly for which no cure currently exists. After over 30 years of intensive research, we have gained extensive knowledge of the genetic and molecular factors involved and their interplay in disease. These findings suggest that different subgroups of AD may exist. Not only are we starting to treat autosomal dominant cases differently from sporadic cases, but we could be observing different underlying pathological mechanisms related to the amyloid cascade hypothesis, immune dysfunction, and a tau-dependent pathology. Genetic, molecular, and, more recently, multi-omic evidence support each of these scenarios, which are highly interconnected but can also point to the different subgroups of AD. The identification of the pathologic triggers and order of events in the disease processes are key to the design of treatments and therapies. Prevention and treatment of AD cannot be attempted using a single approach; different therapeutic strategies at specific disease stages may be appropriate. For successful prevention and treatment, biomarker assays must be designed so that patients can be more accurately monitored at specific points during the course of the disease and potential treatment. In addition, to advance the development of therapeutic drugs, models that better mimic the complexity of the human brain are needed; there have been several advances in this arena. Here, we review significant, recent developments in genetics, omics, and molecular studies that have contributed to the understanding of this disease. We also discuss the implications that these contributions have on medicine.
Keywords: APOE; Alzheimer disease; OMICS; TREM2; amyloid β; biomarkers; neuroinflammation; tau; therapeutics.
Conflict of interest statement
C.C. receives research support from: Biogen, EISAI, Alector, and Parabon. The funders of the study had no role in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. C.C. is a member of the advisory board of Vivid Genetics, Halia Therapeutics, and ADx Healthcare.
Figures
References
-
- Alzheimer’s Association . Alzheimer’s & Dementia. Alzheimer’s Association; Chicago, IL, USA: 2019. 2019 Alzheimer’s Disease Facts and Figures; p. 87.
-
- Lambert J.C., Ibrahim-Verbaas C.A., Harold D., Naj A.C., Sims R., Bellenguez C., DeStafano A.L., Bis J.C., Beecham G.W., Grenier-Boley B., et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. - DOI - PMC - PubMed
-
- Sims R., Ibrahim-Verbaas C.A., Harold D., Naj A.C., Sims R., Bellenguez C., DeStafano A.L., Bis J.C., Beecham G.W., Grenier-Boley B., et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 2017;49:1373–1384. doi: 10.1038/ng.3916. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
