

Reviewing the Significance of Blood-Brain Barrier Disruption in Multiple Sclerosis Pathology and Treatment
- PMID: 34445097
- PMCID: PMC8395058
- DOI: 10.3390/ijms22168370
Reviewing the Significance of Blood-Brain Barrier Disruption in Multiple Sclerosis Pathology and Treatment
Abstract
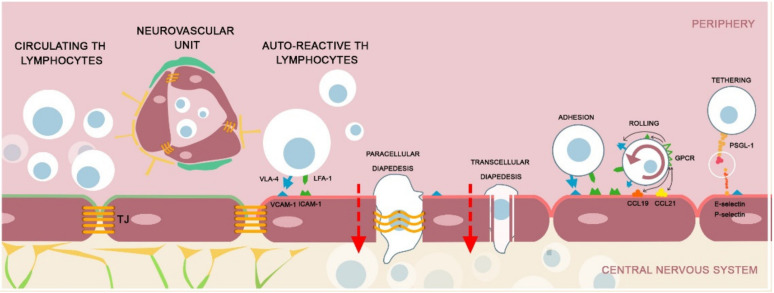
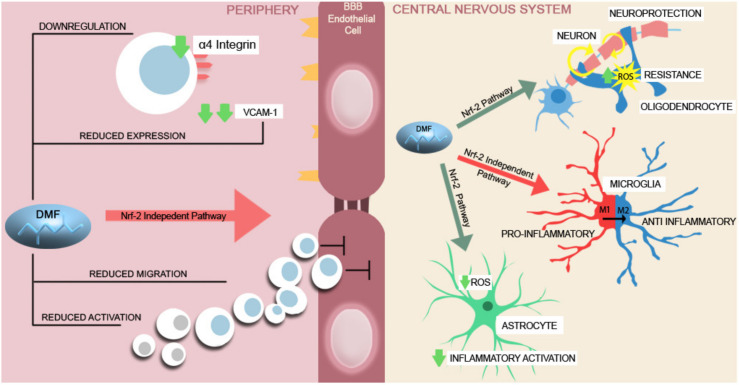
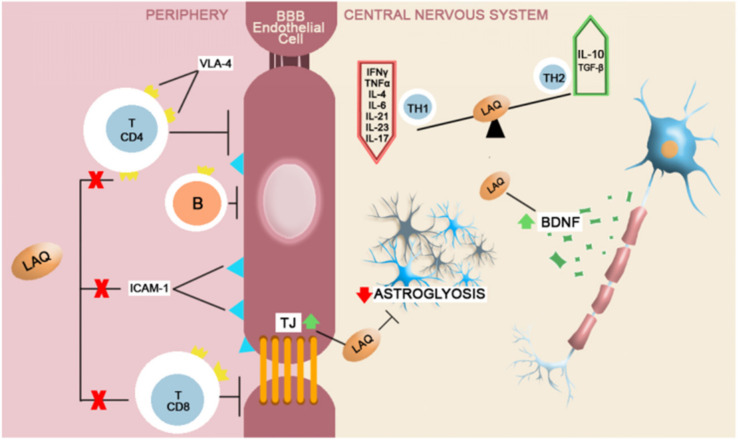
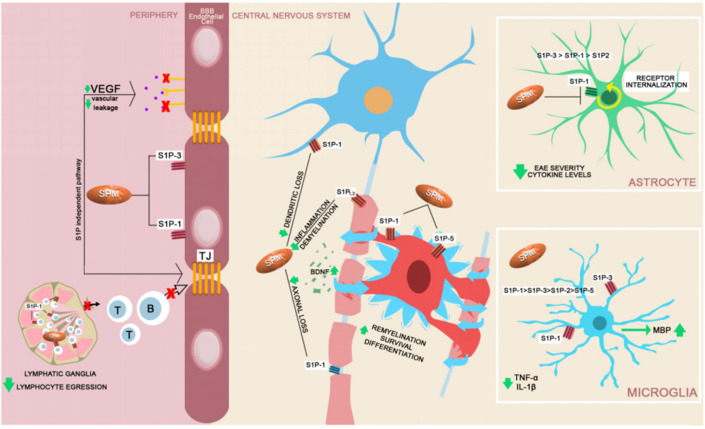
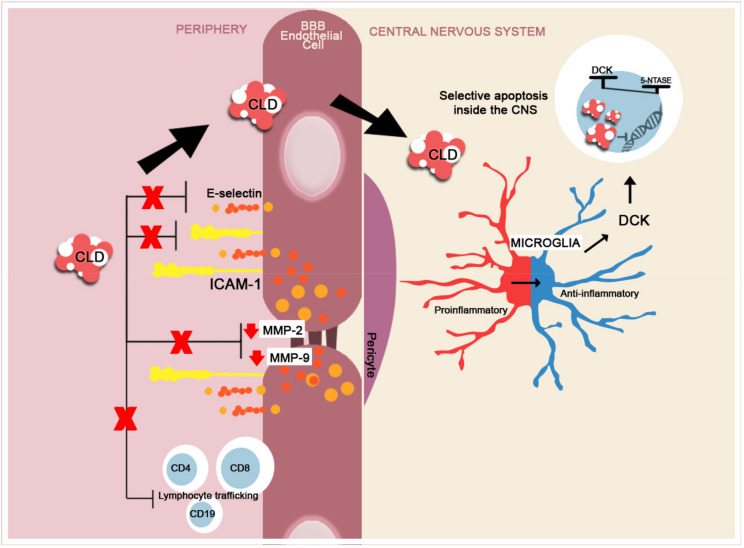
The disruption of blood-brain barrier (BBB) for multiple sclerosis (MS) pathogenesis has a double effect: early on during the onset of the immune attack and later for the CNS self-sustained 'inside-out' demyelination and neurodegeneration processes. This review presents the characteristics of BBB malfunction in MS but mostly highlights current developments regarding the impairment of the neurovascular unit (NVU) and the metabolic and mitochondrial dysfunctions of the BBB's endothelial cells. The hypoxic hypothesis is largely studied and agreed upon recently in the pathologic processes in MS. Hypoxia in MS might be produced per se by the NVU malfunction or secondary to mitochondria dysfunction. We present three different but related terms that denominate the ongoing neurodegenerative process in progressive forms of MS that are indirectly related to BBB disruption: progression independent of relapses, no evidence of disease activity and smoldering demyelination or silent progression. Dimethyl fumarate (DMF), modulators of S1P receptor, cladribine and laquinimode are DMTs that are able to cross the BBB and exhibit beneficial direct effects in the CNS with very different mechanisms of action, providing hope that a combined therapy might be effective in treating MS. Detailed mechanisms of action of these DMTs are described and also illustrated in dedicated images. With increasing knowledge about the involvement of BBB in MS pathology, BBB might become a therapeutic target in MS not only to make it impenetrable against activated immune cells but also to allow molecules that have a neuroprotective effect in reaching the cell target inside the CNS.
Keywords: blood-brain barrier; disease modifying therapies progression; impermeability; multiple sclerosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
