

Insulin Signal Transduction Perturbations in Insulin Resistance
- PMID: 34445300
- PMCID: PMC8395322
- DOI: 10.3390/ijms22168590
Insulin Signal Transduction Perturbations in Insulin Resistance
Abstract
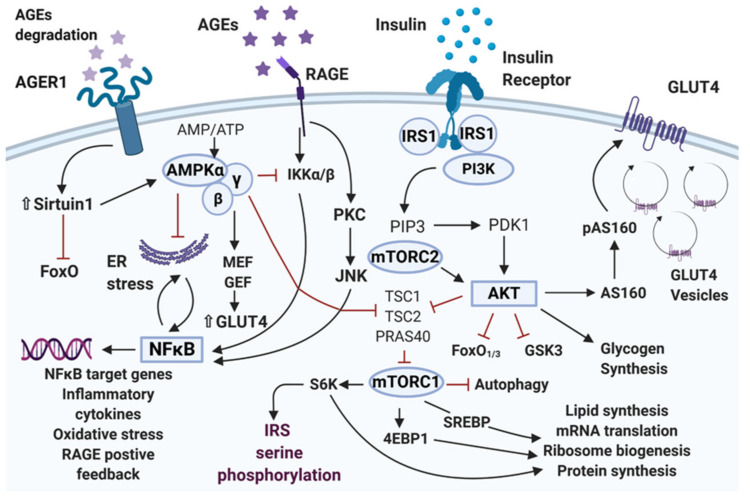
Type 2 diabetes mellitus is a widespread medical condition, characterized by high blood glucose and inadequate insulin action, which leads to insulin resistance. Insulin resistance in insulin-responsive tissues precedes the onset of pancreatic β-cell dysfunction. Multiple molecular and pathophysiological mechanisms are involved in insulin resistance. Insulin resistance is a consequence of a complex combination of metabolic disorders, lipotoxicity, glucotoxicity, and inflammation. There is ample evidence linking different mechanistic approaches as the cause of insulin resistance, but no central mechanism is yet described as an underlying reason behind this condition. This review combines and interlinks the defects in the insulin signal transduction pathway of the insulin resistance state with special emphasis on the AGE-RAGE-NF-κB axis. Here, we describe important factors that play a crucial role in the pathogenesis of insulin resistance to provide directionality for the events. The interplay of inflammation and oxidative stress that leads to β-cell decline through the IAPP-RAGE induced β-cell toxicity is also addressed. Overall, by generating a comprehensive overview of the plethora of mechanisms involved in insulin resistance, we focus on the establishment of unifying mechanisms to provide new insights for the future interventions of type 2 diabetes mellitus.
Keywords: hyperglycemia; insulin resistance; insulin signaling; pancreatic beta cells; type 2 diabetes mellitus.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
References
-
- Barnett D.M., Krall L.P. Joslin’s Diabetes Mellitus. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2005. The history of diabetes; pp. 1–17.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
