

Technology dictates algorithms: recent developments in read alignment
- PMID: 34446078
- PMCID: PMC8390189
- DOI: 10.1186/s13059-021-02443-7
Technology dictates algorithms: recent developments in read alignment
Abstract
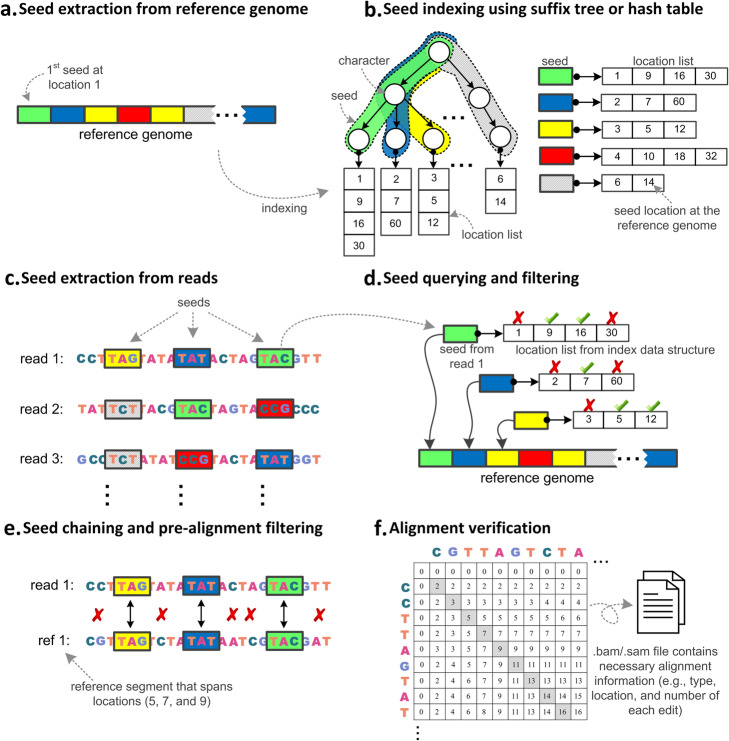
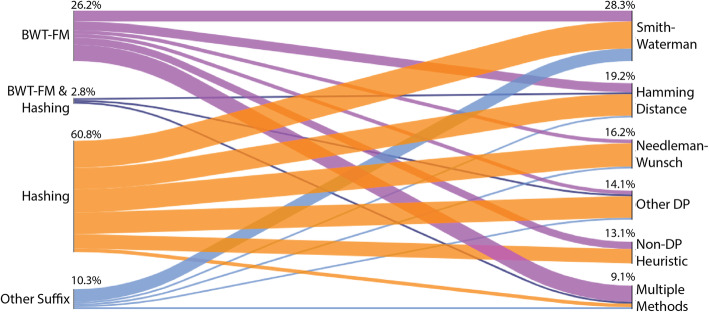
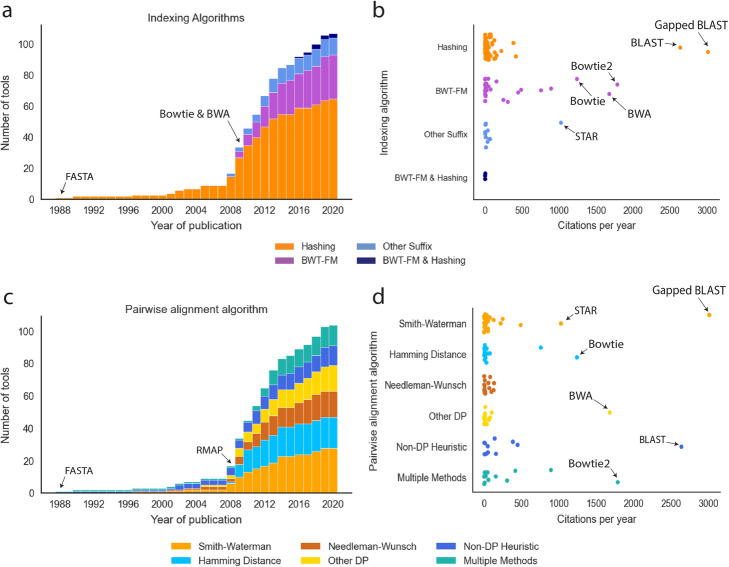
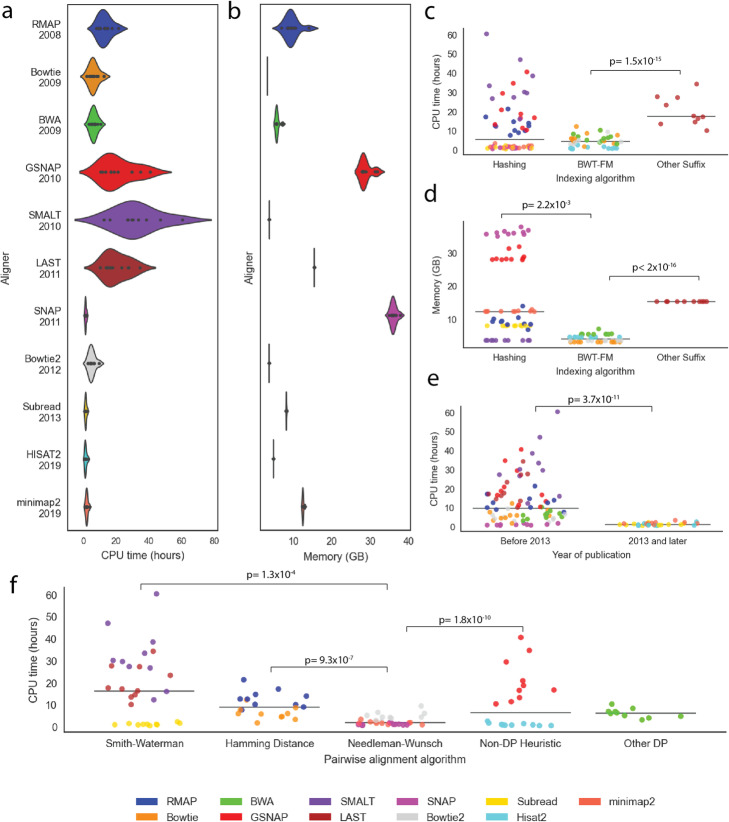
Aligning sequencing reads onto a reference is an essential step of the majority of genomic analysis pipelines. Computational algorithms for read alignment have evolved in accordance with technological advances, leading to today's diverse array of alignment methods. We provide a systematic survey of algorithmic foundations and methodologies across 107 alignment methods, for both short and long reads. We provide a rigorous experimental evaluation of 11 read aligners to demonstrate the effect of these underlying algorithms on speed and efficiency of read alignment. We discuss how general alignment algorithms have been tailored to the specific needs of various domains in biology.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Weissenbach J. The Human Genome. 2002. Human Genome Project: Past, Present, Future; pp. 1–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
