

Primary Immune Regulatory Disorders With an Autoimmune Lymphoproliferative Syndrome-Like Phenotype: Immunologic Evaluation, Early Diagnosis and Management
- PMID: 34447369
- PMCID: PMC8382720
- DOI: 10.3389/fimmu.2021.671755
Primary Immune Regulatory Disorders With an Autoimmune Lymphoproliferative Syndrome-Like Phenotype: Immunologic Evaluation, Early Diagnosis and Management
Abstract
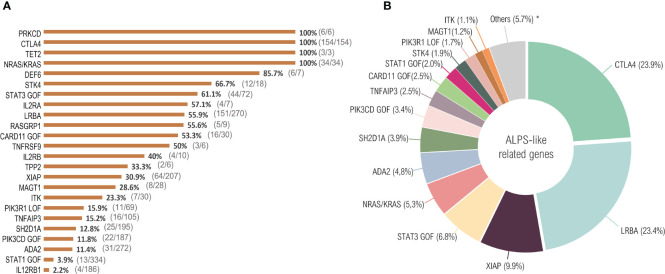
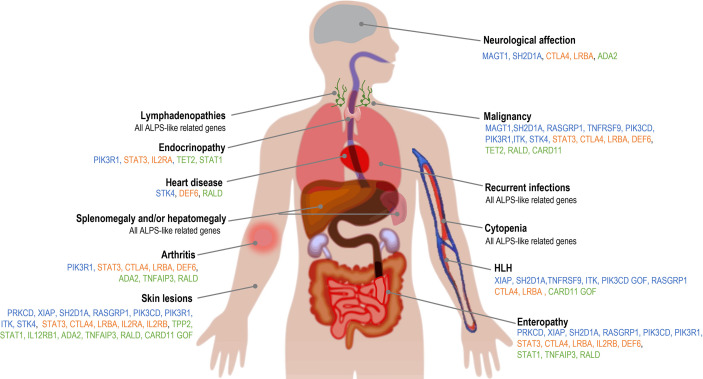
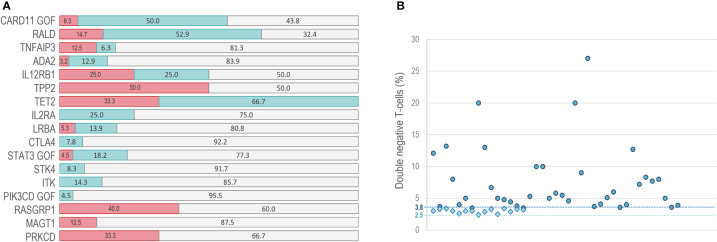
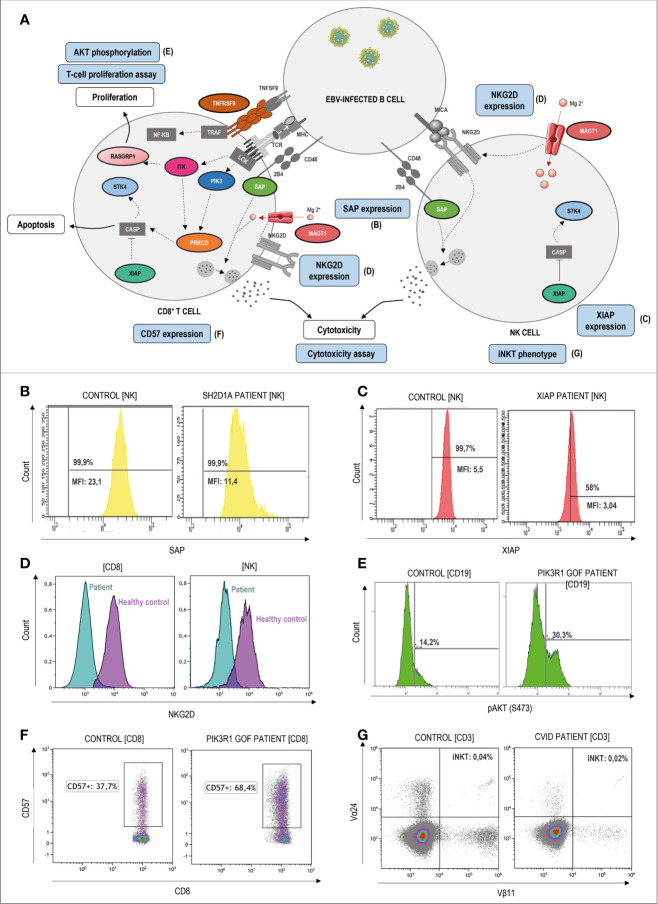
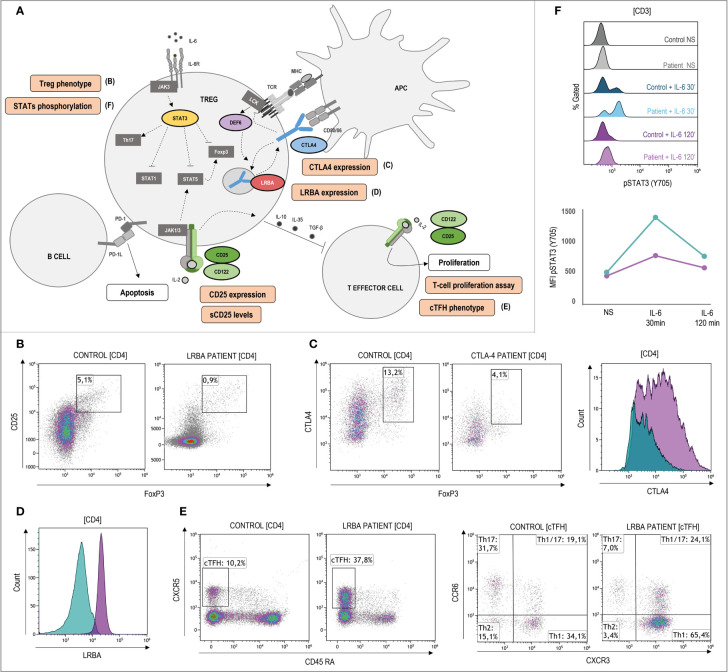
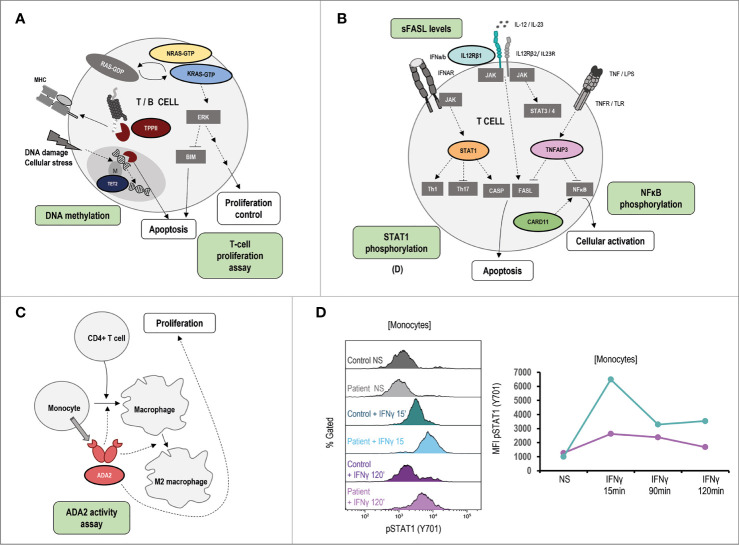
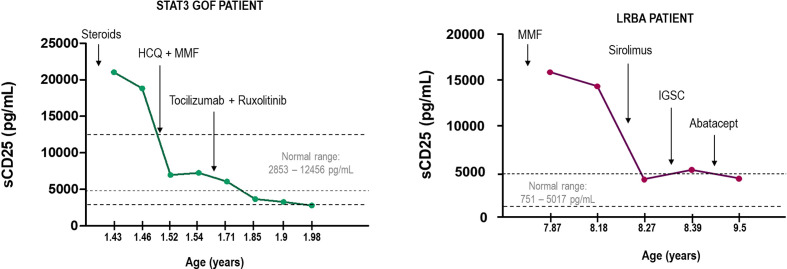
Primary immune regulatory disorders (PIRD) are associated with autoimmunity, autoinflammation and/or dysregulation of lymphocyte homeostasis. Autoimmune lymphoproliferative syndrome (ALPS) is a PIRD due to an apoptotic defect in Fas-FasL pathway and characterized by benign and chronic lymphoproliferation, autoimmunity and increased risk of lymphoma. Clinical manifestations and typical laboratory biomarkers of ALPS have also been found in patients with a gene defect out of the Fas-FasL pathway (ALPS-like disorders). Following the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA), we identified more than 600 patients suffering from 24 distinct genetic defects described in the literature with an autoimmune lymphoproliferative phenotype (ALPS-like syndromes) corresponding to phenocopies of primary immunodeficiency (PID) (NRAS, KRAS), susceptibility to EBV (MAGT1, PRKCD, XIAP, SH2D1A, RASGRP1, TNFRSF9), antibody deficiency (PIK3CD gain of function (GOF), PIK3R1 loss of function (LOF), CARD11 GOF), regulatory T-cells defects (CTLA4, LRBA, STAT3 GOF, IL2RA, IL2RB, DEF6), combined immunodeficiencies (ITK, STK4), defects in intrinsic and innate immunity and predisposition to infection (STAT1 GOF, IL12RB1) and autoimmunity/autoinflammation (ADA2, TNFAIP3,TPP2, TET2). CTLA4 and LRBA patients correspond around to 50% of total ALPS-like cases. However, only 100% of CTLA4, PRKCD, TET2 and NRAS/KRAS reported patients had an ALPS-like presentation, while the autoimmunity and lymphoproliferation combination resulted rare in other genetic defects. Recurrent infections, skin lesions, enteropathy and malignancy are the most common clinical manifestations. Some approaches available for the immunological study and identification of ALPS-like patients through flow cytometry and ALPS biomarkers are provided in this work. Protein expression assays for NKG2D, XIAP, SAP, CTLA4 and LRBA deficiencies and functional studies of AKT, STAT1 and STAT3 phosphorylation, are showed as useful tests. Patients suspected to suffer from one of these disorders require rapid and correct diagnosis allowing initiation of tailored specific therapeutic strategies and monitoring thereby improving the prognosis and their quality of life.
Keywords: ALPS; ALPS-like; autoimmunity; immune dysregulation; lymphoproliferation; malignancy.
Copyright © 2021 López-Nevado, González-Granado, Ruiz-García, Pleguezuelo, Cabrera-Marante, Salmón, Blanco-Lobo, Domínguez-Pinilla, Rodríguez-Pena, Sebastián, Cruz-Rojo, Olbrich, Ruiz-Contreras, Paz-Artal, Neth and Allende.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. . Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. 10.1007/s10875-019-00737-x - DOI - PMC - PubMed