

Interaction of Poliovirus Capsid Proteins with the Cellular Autophagy Pathway
- PMID: 34452452
- PMCID: PMC8402707
- DOI: 10.3390/v13081587
Interaction of Poliovirus Capsid Proteins with the Cellular Autophagy Pathway
Abstract
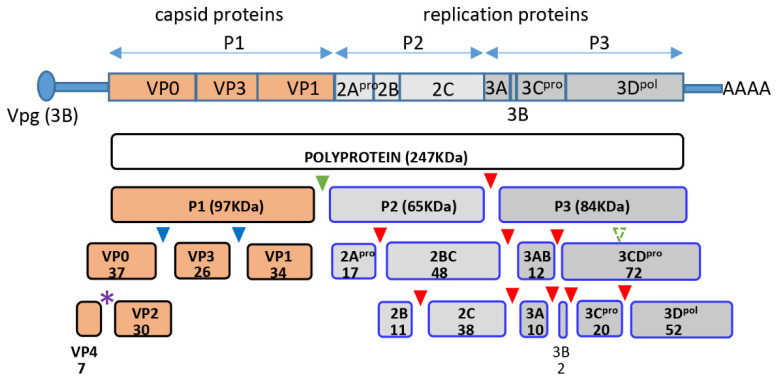
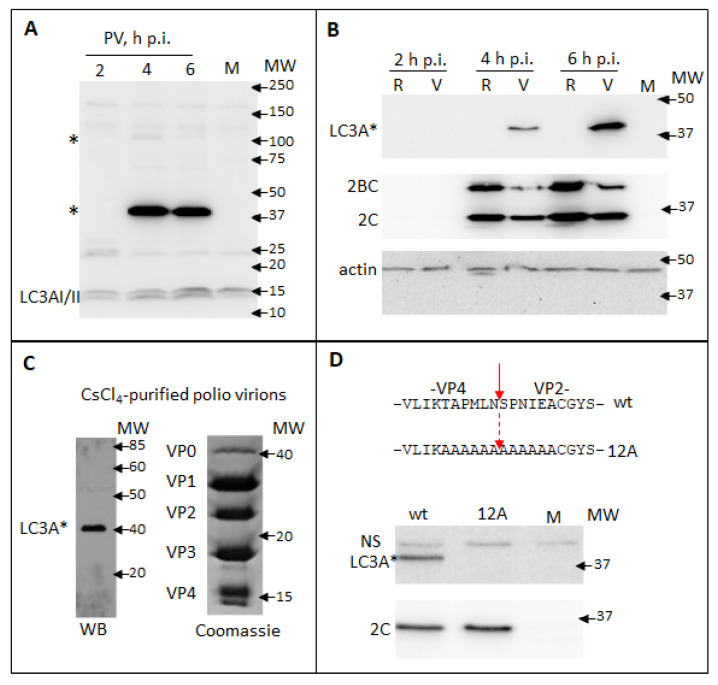
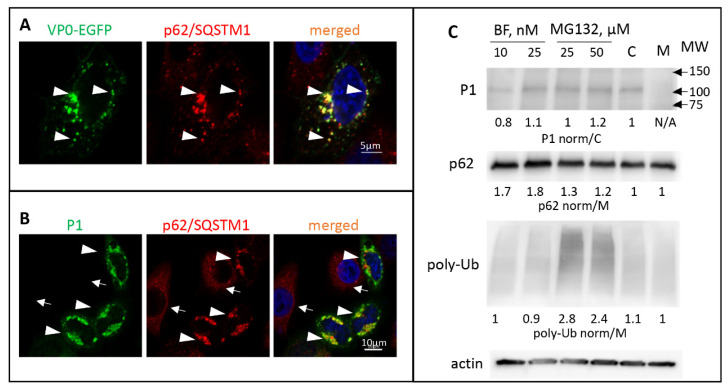
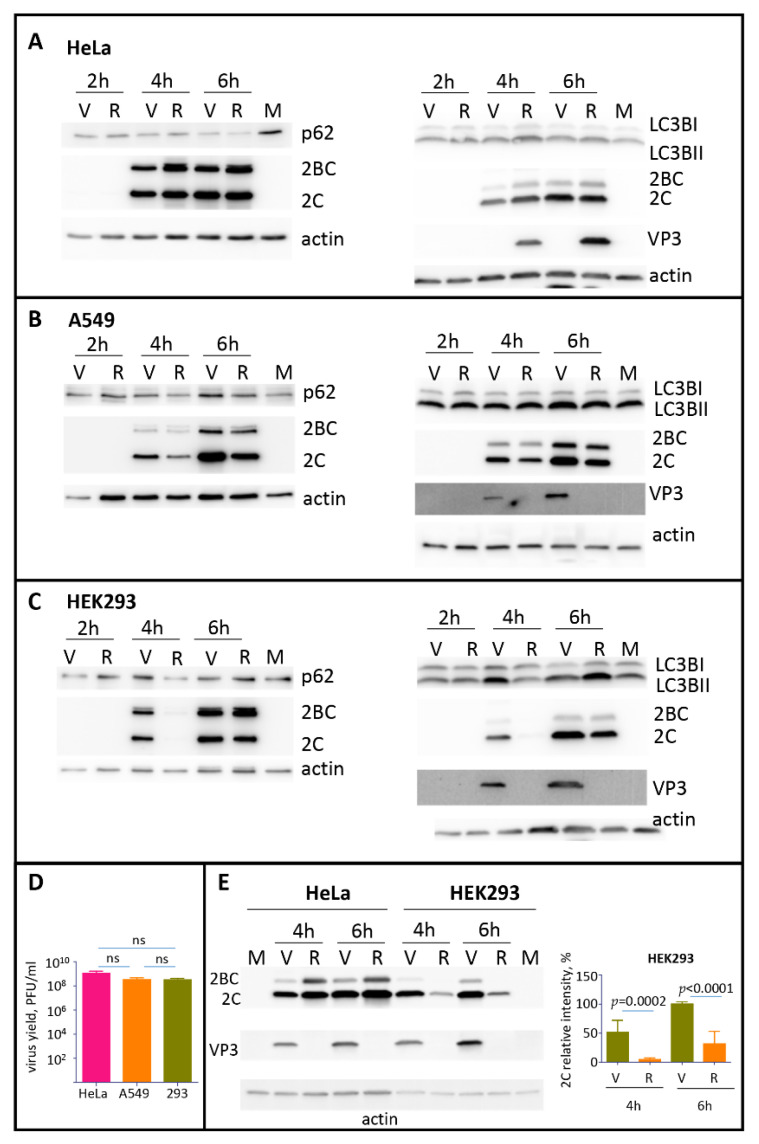
The capsid precursor P1 constitutes the N-terminal part of the enterovirus polyprotein. It is processed into VP0, VP3, and VP1 by the viral proteases, and VP0 is cleaved autocatalytically into VP4 and VP2. We observed that poliovirus VP0 is recognized by an antibody against a cellular autophagy protein, LC3A. The LC3A-like epitope overlapped the VP4/VP2 cleavage site. Individually expressed VP0-EGFP and P1 strongly colocalized with a marker of selective autophagy, p62/SQSTM1. To assess the role of capsid proteins in autophagy development we infected different cells with poliovirus or encapsidated polio replicon coding for only the replication proteins. We analyzed the processing of LC3B and p62/SQSTM1, markers of the initiation and completion of the autophagy pathway and investigated the association of the viral antigens with these autophagy proteins in infected cells. We observed cell-type-specific development of autophagy upon infection and found that only the virion signal strongly colocalized with p62/SQSTM1 early in infection. Collectively, our data suggest that activation of autophagy is not required for replication, and that capsid proteins contain determinants targeting them to p62/SQSTM1-dependent sequestration. Such a strategy may control the level of capsid proteins so that viral RNAs are not removed from the replication/translation pool prematurely.
Keywords: LC3 processing; autophagy; enterovirus replication; enteroviruses; poliovirus.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
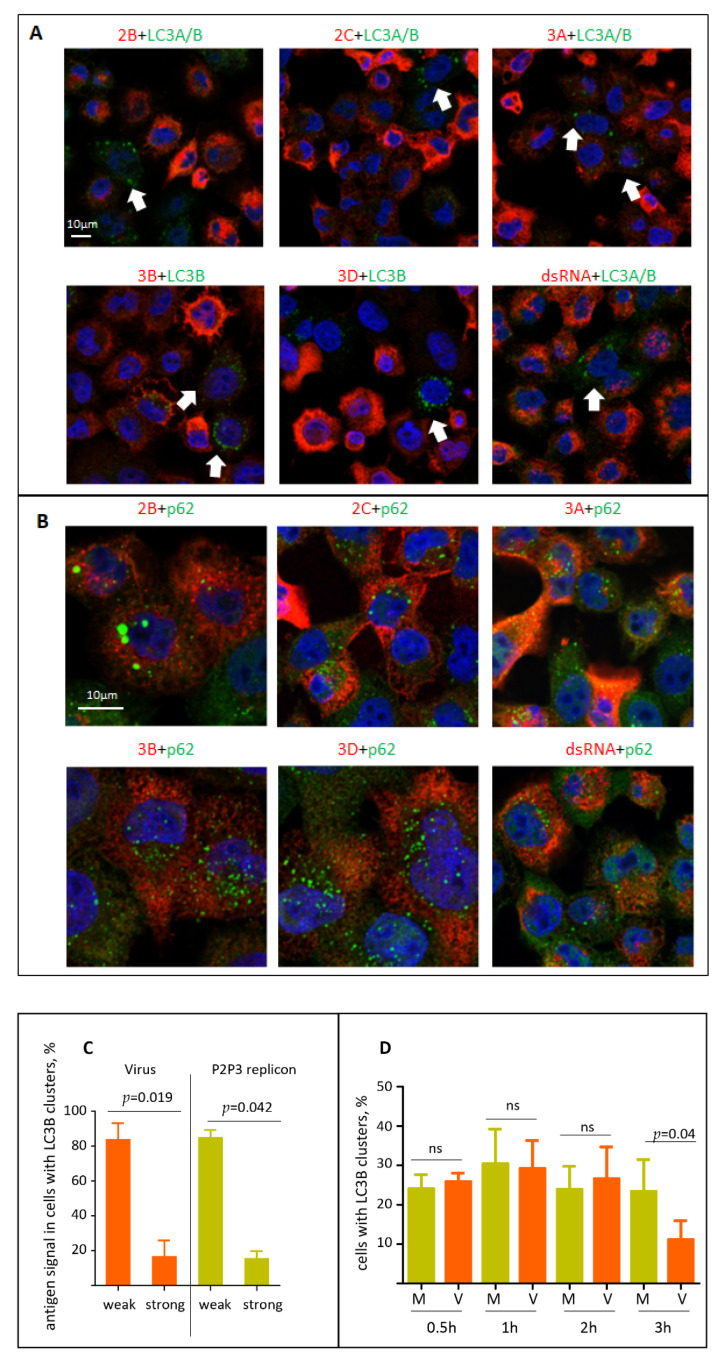
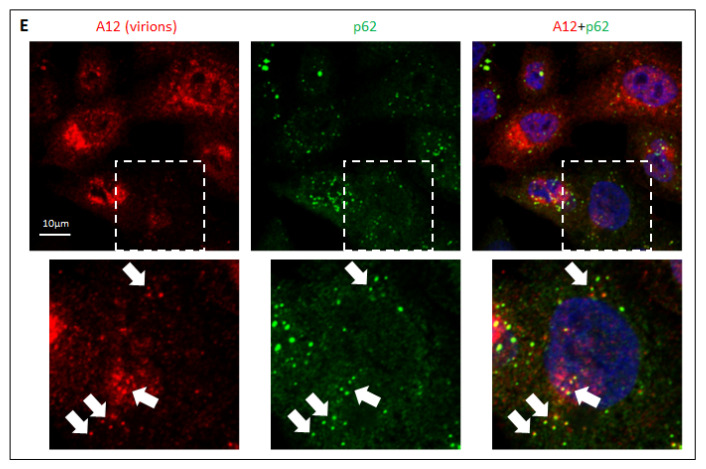
References
-
- Fields B.N., Knipe D.M., Howley P.M. Fields Virology. 5th ed. Wolters Kluwer Health/Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2007.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
