

Dual-mechanism estrogen receptor inhibitors
- PMID: 34452998
- PMCID: PMC8536401
- DOI: 10.1073/pnas.2101657118
Dual-mechanism estrogen receptor inhibitors
Abstract
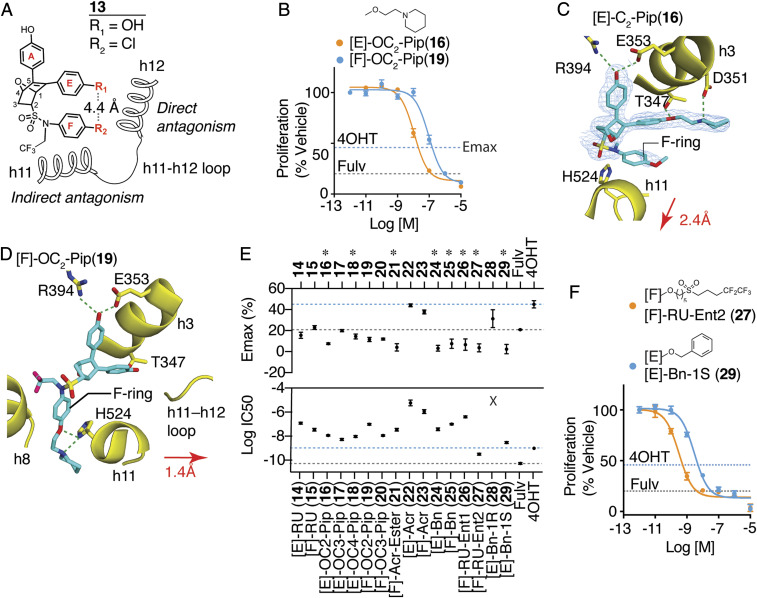
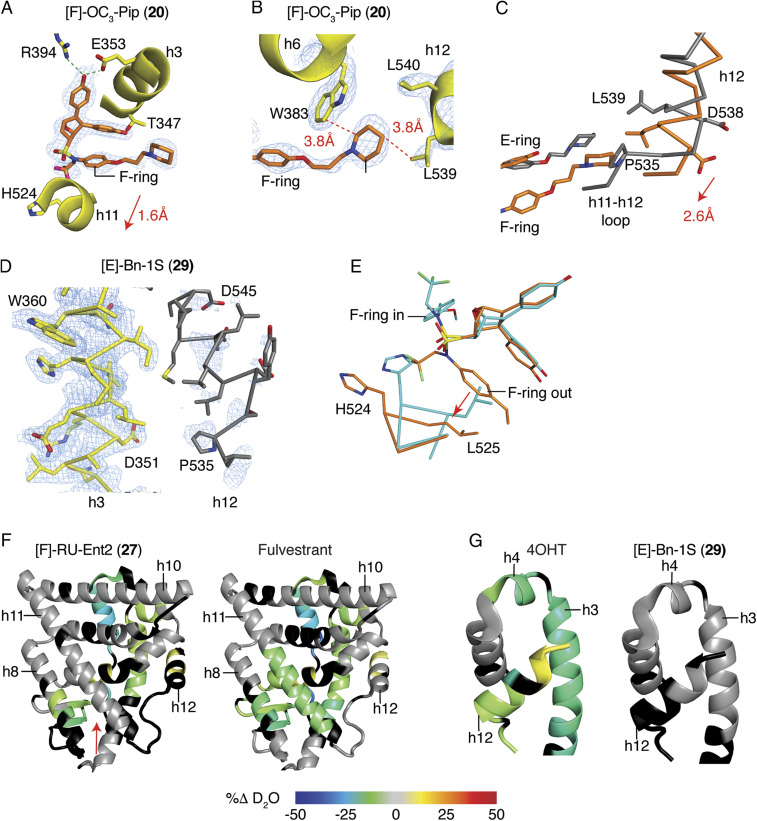
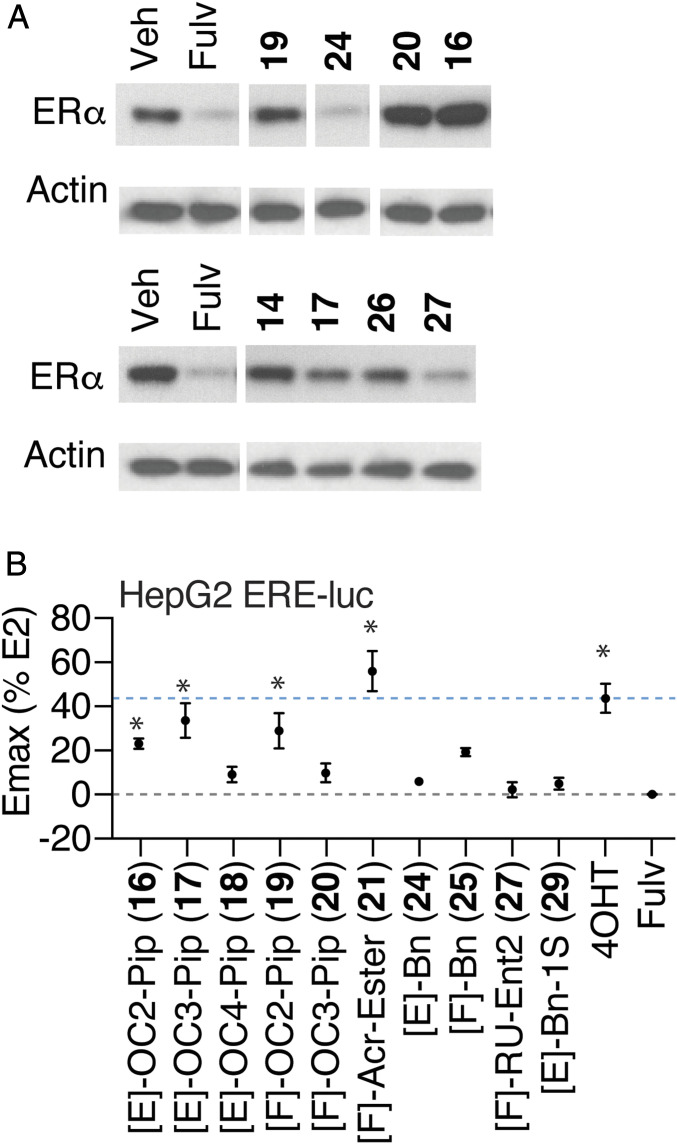
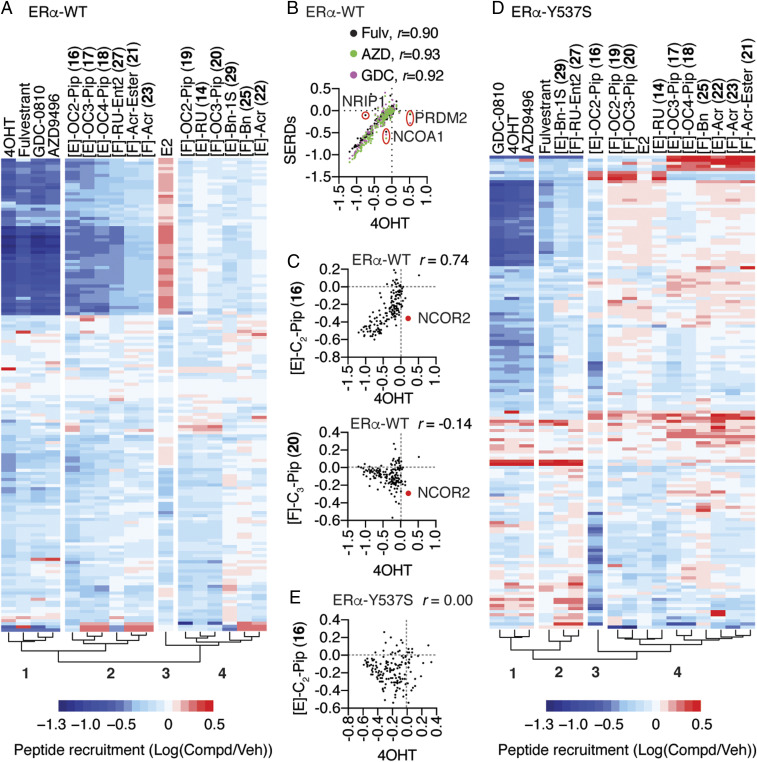
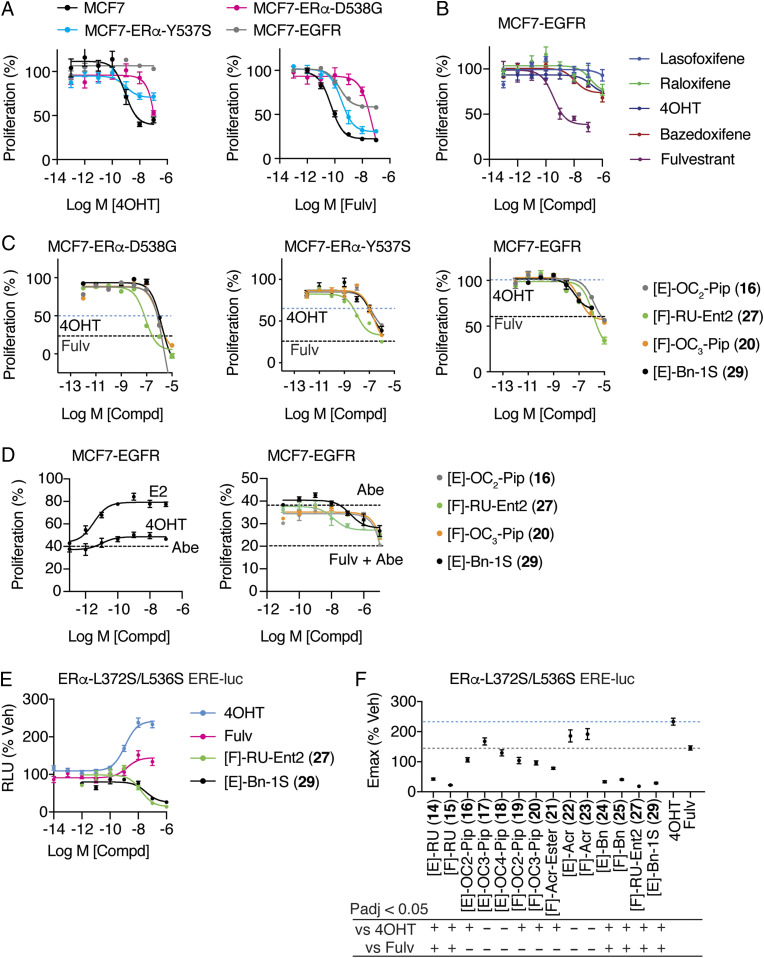
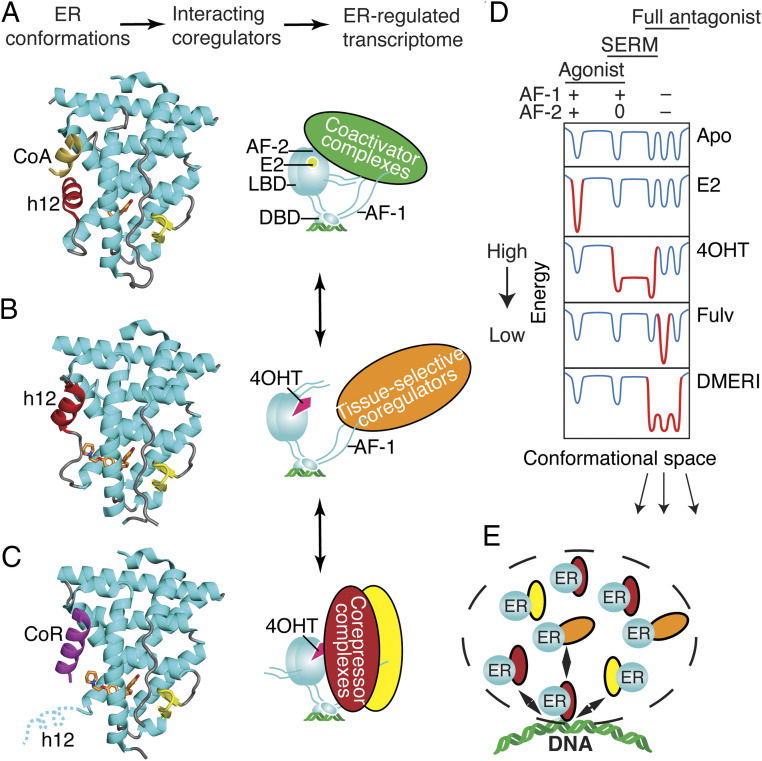
Efforts to improve estrogen receptor-α (ER)-targeted therapies in breast cancer have relied upon a single mechanism, with ligands having a single side chain on the ligand core that extends outward to determine antagonism of breast cancer growth. Here, we describe inhibitors with two ER-targeting moieties, one of which uses an alternate structural mechanism to generate full antagonism, freeing the side chain to independently determine other critical properties of the ligands. By combining two molecular targeting approaches into a single ER ligand, we have generated antiestrogens that function through new mechanisms and structural paradigms to achieve antagonism. These dual-mechanism ER inhibitors (DMERIs) cause alternate, noncanonical structural perturbations of the receptor ligand-binding domain (LBD) to antagonize proliferation in ER-positive breast cancer cells and in allele-specific resistance models. Our structural analyses with DMERIs highlight marked differences from current standard-of-care, single-mechanism antiestrogens. These findings uncover an enhanced flexibility of the ER LBD through which it can access nonconsensus conformational modes in response to DMERI binding, broadly and effectively suppressing ER activity.
Keywords: SERM; X-ray crystallography; breast cancer; cancer therapy; estrogen receptor.
Conflict of interest statement
Competing interest statement: J.A.K. is a founder and stockholder of Radius Health Inc. and a consultant of Celcuity Inc. B.S.K. is a consultant of Celcuity Inc.
Figures
References
-
- Rugo H. S., et al., Endocrine therapy for hormone receptor-positive metastatic breast cancer: American Society of Clinical Oncology guideline. J. Clin. Oncol. 34, 3069–3103 (2016). - PubMed
-
- Tryfonidis K., Zardavas D., Katzenellenbogen B. S., Piccart M., Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat. Rev. 50, 68–81 (2016). - PubMed
-
- Lerner H. J., Band P. R., Israel L., Leung B. S., Phase II study of tamoxifen: Report of 74 patients with stage IV breast cancer. Cancer Treat. Rep. 60, 1431–1435 (1976). - PubMed
-
- Jordan V. C., Phelps E., Lindgren J. U., Effects of anti-estrogens on bone in castrated and intact female rats. Breast Cancer Res. Treat. 10, 31–35 (1987). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
