

CTCF and transcription influence chromatin structure re-configuration after mitosis
- PMID: 34453048
- PMCID: PMC8397779
- DOI: 10.1038/s41467-021-25418-5
CTCF and transcription influence chromatin structure re-configuration after mitosis
Abstract
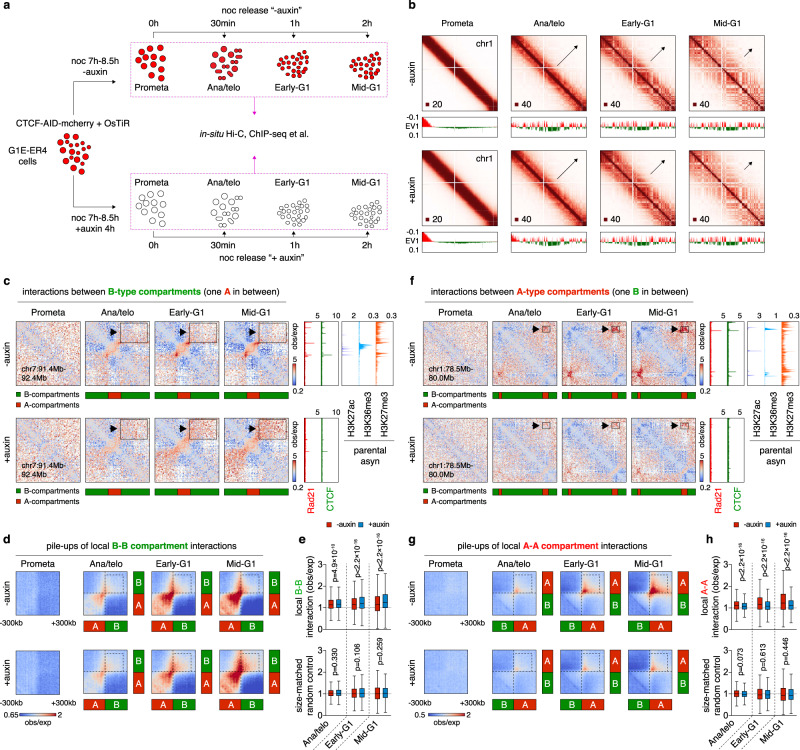
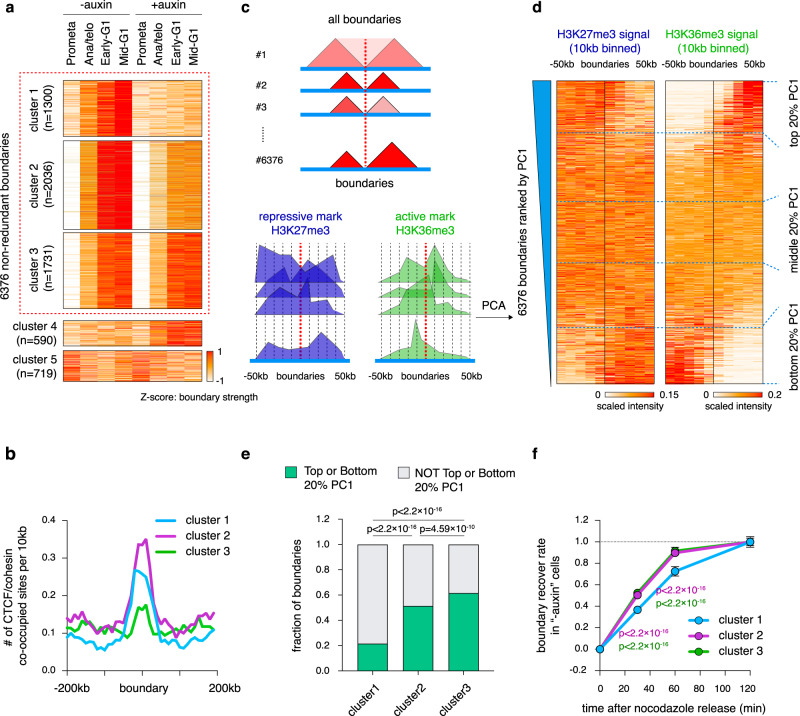
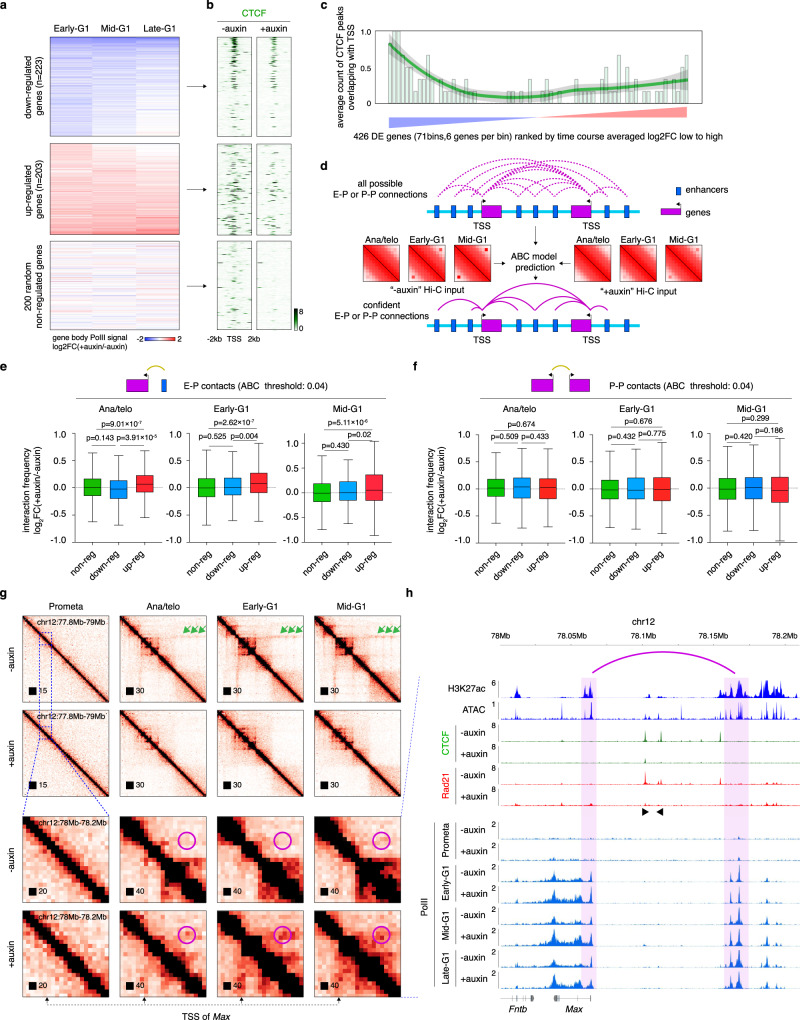
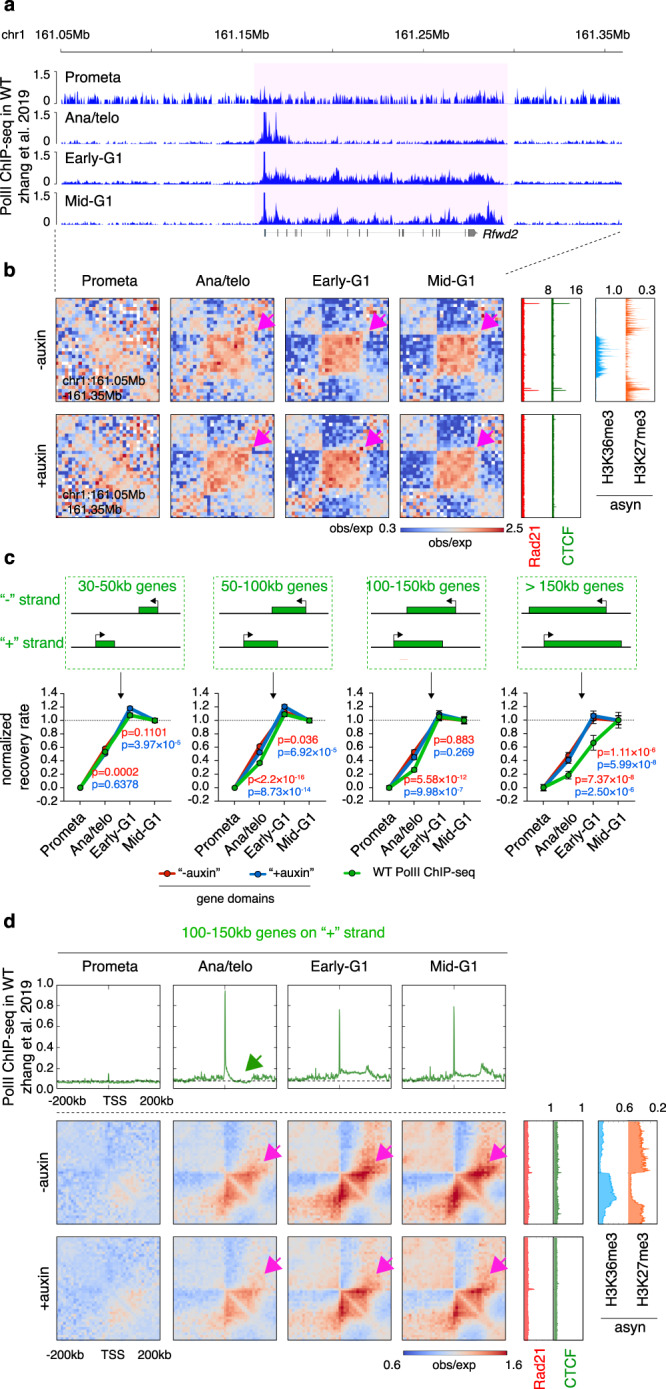
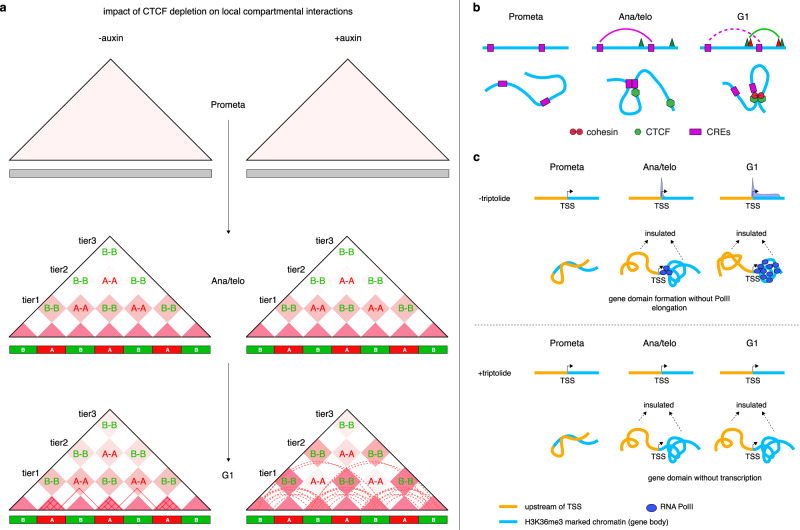
During mitosis, transcription is globally attenuated and chromatin architecture is dramatically reconfigured. We exploited the M- to G1-phase progression to interrogate the contributions of the architectural factor CTCF and the process of transcription to genome re-sculpting in newborn nuclei. Depletion of CTCF during the M- to G1-phase transition alters short-range compartmentalization after mitosis. Chromatin domain boundary re-formation is impaired upon CTCF loss, but a subset of boundaries, characterized by transitions in chromatin states, is established normally. Without CTCF, structural loops fail to form, leading to illegitimate contacts between cis-regulatory elements (CREs). Transient CRE contacts that are normally resolved after telophase persist deeply into G1-phase in CTCF-depleted cells. CTCF loss-associated gains in transcription are often linked to increased, normally illegitimate enhancer-promoter contacts. In contrast, at genes whose expression declines upon CTCF loss, CTCF seems to function as a conventional transcription activator, independent of its architectural role. CTCF-anchored structural loops facilitate formation of CRE loops nested within them, especially those involving weak CREs. Transcription inhibition does not significantly affect global architecture or transcription start site-associated boundaries. However, ongoing transcription contributes considerably to the formation of gene domains, regions of enriched contacts along gene bodies. Notably, gene domains emerge in ana/telophase prior to completion of the first round of transcription, suggesting that epigenetic features in gene bodies contribute to genome reconfiguration prior to transcription. The focus on the de novo formation of nuclear architecture during G1 entry yields insights into the contributions of CTCF and transcription to chromatin architecture dynamics during the mitosis to G1-phase progression.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
