

The plasticity of pancreatic cancer stem cells: implications in therapeutic resistance
- PMID: 34453639
- PMCID: PMC8556195
- DOI: 10.1007/s10555-021-09979-x
The plasticity of pancreatic cancer stem cells: implications in therapeutic resistance
Abstract
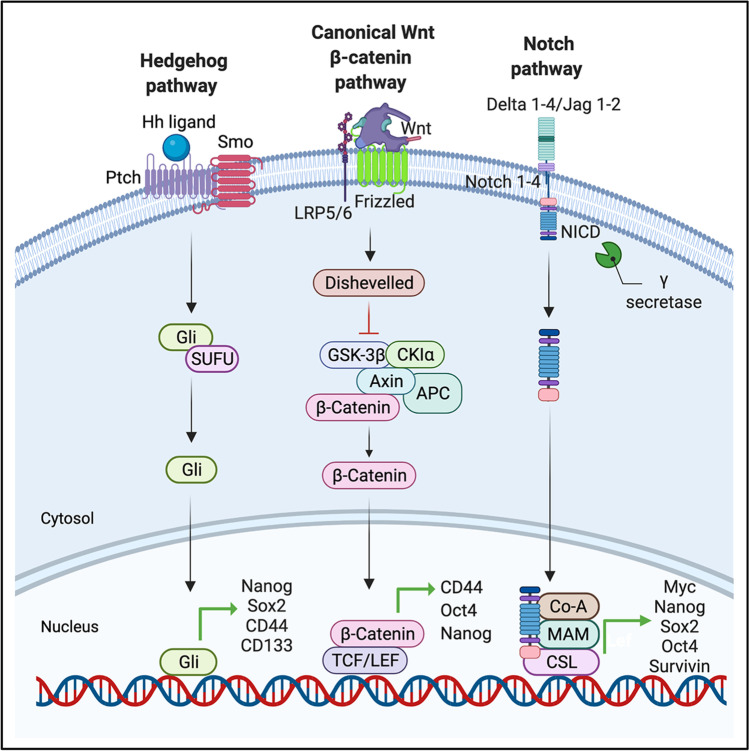
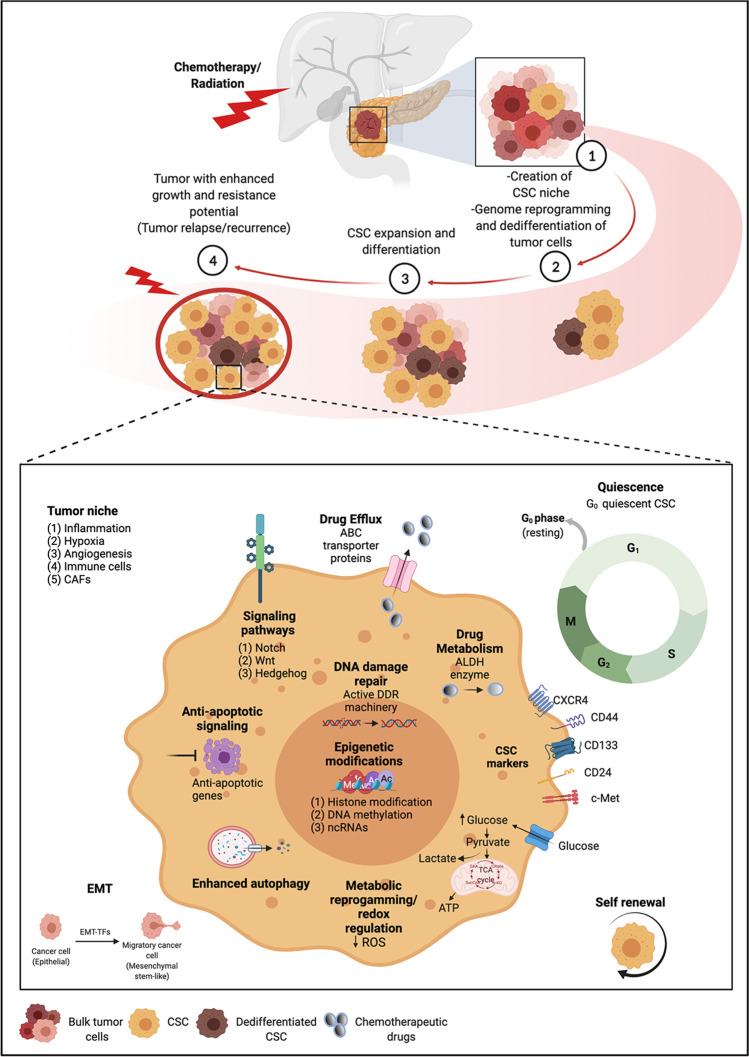
The ever-growing perception of cancer stem cells (CSCs) as a plastic state rather than a hardwired defined entity has evolved our understanding of the functional and biological plasticity of these elusive components in malignancies. Pancreatic cancer (PC), based on its biological features and clinical evolution, is a prototypical example of a CSC-driven disease. Since the discovery of pancreatic CSCs (PCSCs) in 2007, evidence has unraveled their control over many facets of the natural history of PC, including primary tumor growth, metastatic progression, disease recurrence, and acquired drug resistance. Consequently, the current near-ubiquitous treatment regimens for PC using aggressive cytotoxic agents, aimed at ''tumor debulking'' rather than eradication of CSCs, have proven ineffective in providing clinically convincing improvements in patients with this dreadful disease. Herein, we review the key hallmarks as well as the intrinsic and extrinsic resistance mechanisms of CSCs that mediate treatment failure in PC and enlist the potential CSC-targeting 'natural agents' that are gaining popularity in recent years. A better understanding of the molecular and functional landscape of PCSC-intrinsic evasion of chemotherapeutic drugs offers a facile opportunity for treating PC, an intractable cancer with a grim prognosis and in dire need of effective therapeutic advances.
Keywords: Cancer stem cells; Drug resistance; Epithelial to mesenchymal transition; Oncogenic signaling; Pancreatic cancer.
© 2021. The Author(s).
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Pancreatic cancer stem cells: A state or an entity?Semin Cancer Biol. 2018 Dec;53:223-231. doi: 10.1016/j.semcancer.2018.08.007. Epub 2018 Aug 18. Semin Cancer Biol. 2018. PMID: 30130664 Review.
-
Pancreatic cancer stem cells: emerging target for designing novel therapy.Cancer Lett. 2013 Sep 10;338(1):94-100. doi: 10.1016/j.canlet.2012.03.018. Epub 2012 Mar 20. Cancer Lett. 2013. PMID: 22445908 Free PMC article. Review.
-
Pancreatic cancer stem cells and EMT in drug resistance and metastasis.Minerva Chir. 2009 Oct;64(5):489-500. Minerva Chir. 2009. PMID: 19859039 Free PMC article. Review.
-
Concise Review: Stem Cells in Pancreatic Cancer: From Concept to Translation.Stem Cells. 2015 Oct;33(10):2893-902. doi: 10.1002/stem.2114. Epub 2015 Aug 14. Stem Cells. 2015. PMID: 26202953 Review.
-
Targeting pancreatic cancer stem cells for cancer therapy.Biochim Biophys Acta. 2012 Dec;1826(2):385-99. doi: 10.1016/j.bbcan.2012.06.002. Epub 2012 Jun 20. Biochim Biophys Acta. 2012. PMID: 22728049 Review.
Cited by
-
Anticancer Activity of Novel Difluorinated Curcumin Analog and Its Inclusion Complex with 2-Hydroxypropyl-β-Cyclodextrin against Pancreatic Cancer.Int J Mol Sci. 2023 Mar 28;24(7):6336. doi: 10.3390/ijms24076336. Int J Mol Sci. 2023. PMID: 37047307 Free PMC article.
-
Induction of Apoptosis in Human Pancreatic Cancer Stem Cells by the Endoplasmic Reticulum-Targeted Alkylphospholipid Analog Edelfosine and Potentiation by Autophagy Inhibition.Cancers (Basel). 2021 Dec 5;13(23):6124. doi: 10.3390/cancers13236124. Cancers (Basel). 2021. PMID: 34885233 Free PMC article.
-
Tumor microenvironment acidosis favors pancreatic cancer stem cell properties and in vivo metastasis.iScience. 2025 Feb 5;28(3):111956. doi: 10.1016/j.isci.2025.111956. eCollection 2025 Mar 21. iScience. 2025. PMID: 40083719 Free PMC article.
-
Ferroptosis- and stemness inhibition-mediated therapeutic potency of ferrous oxide nanoparticles-diethyldithiocarbamate using a co-spheroid 3D model of pancreatic cancer.J Gastroenterol. 2025 May;60(5):641-657. doi: 10.1007/s00535-025-02213-3. Epub 2025 Jan 31. J Gastroenterol. 2025. PMID: 39888413 Free PMC article.
-
The strategies to cure cancer patients by eradicating cancer stem-like cells.Mol Cancer. 2023 Oct 18;22(1):171. doi: 10.1186/s12943-023-01867-y. Mol Cancer. 2023. PMID: 37853413 Free PMC article. Review.
References
-
- Cancer Stat Facts: Pancreatic Cancer. Surveillance, Epidemiology, and End Results Program; Available from: https://seer.cancer.gov/statfacts/html/pancreas.html. Accessed 20 May 2021.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
