

Phylogenetic reconstruction of the initial stages of the spread of the SARS-CoV-2 virus in the Eurasian and American continents by analyzing genomic data
- PMID: 34454972
- PMCID: PMC8388146
- DOI: 10.1016/j.virusres.2021.198551
Phylogenetic reconstruction of the initial stages of the spread of the SARS-CoV-2 virus in the Eurasian and American continents by analyzing genomic data
Abstract
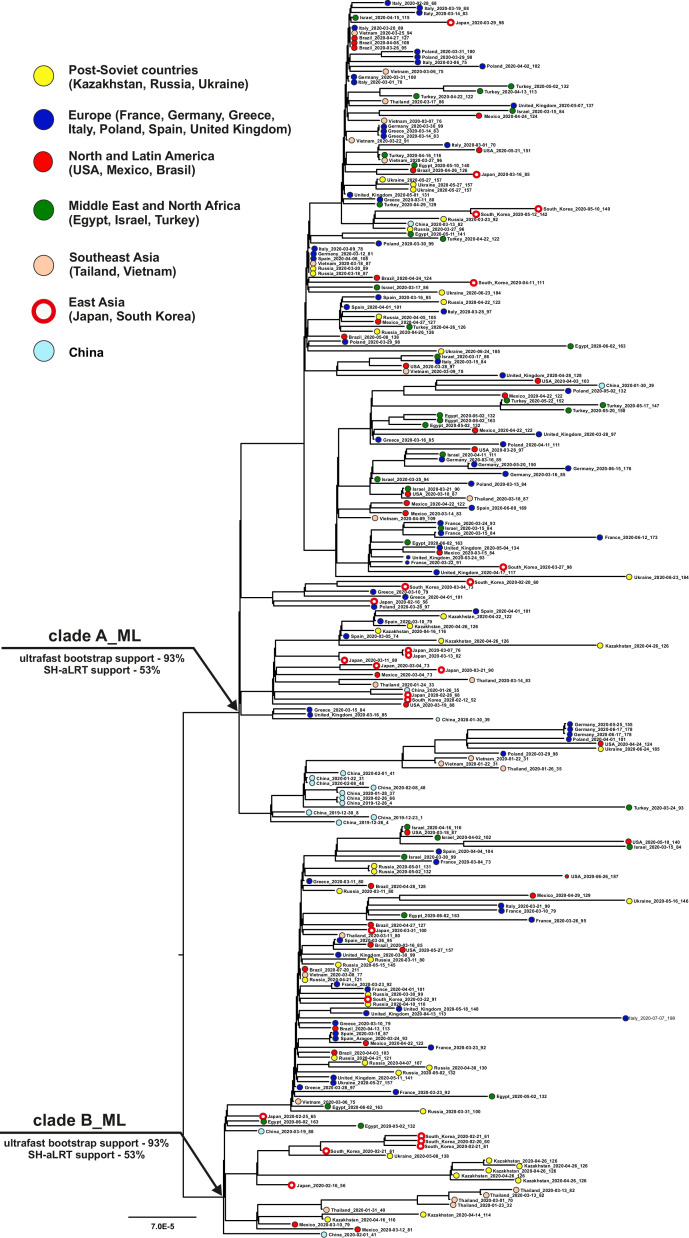
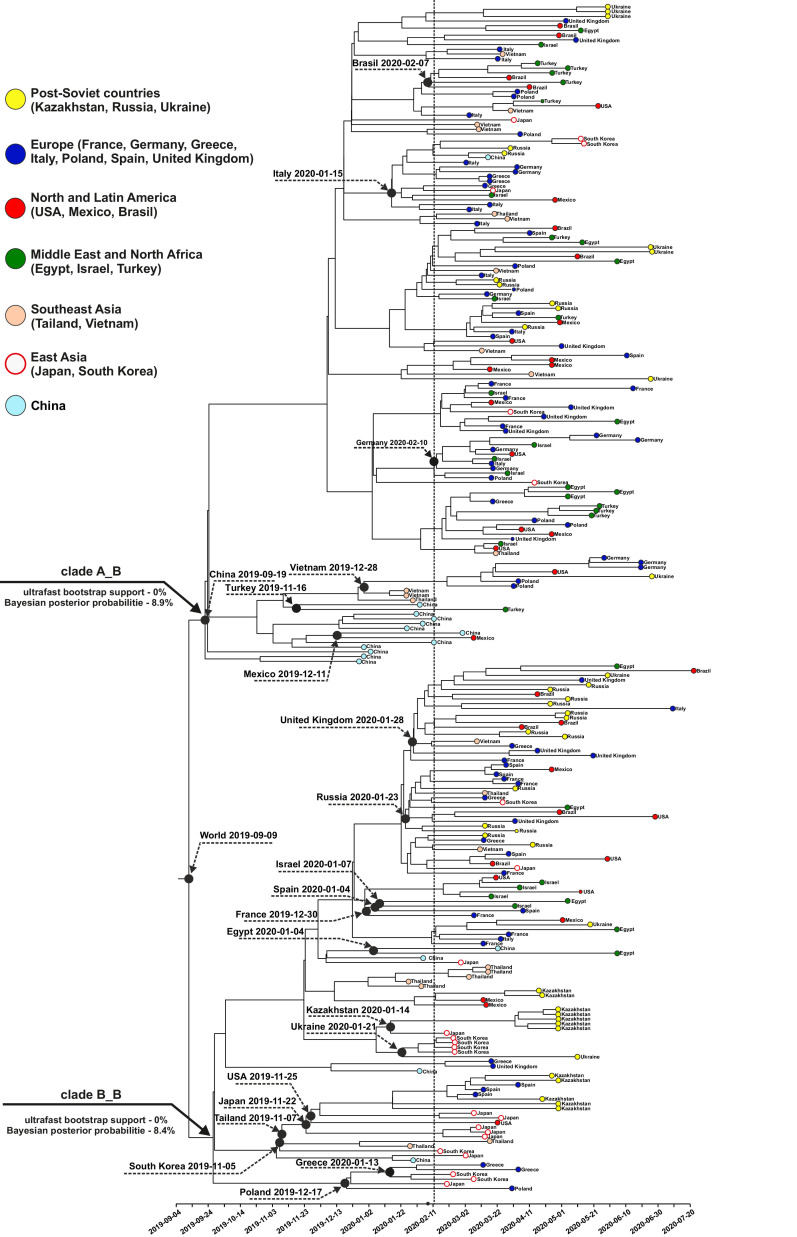
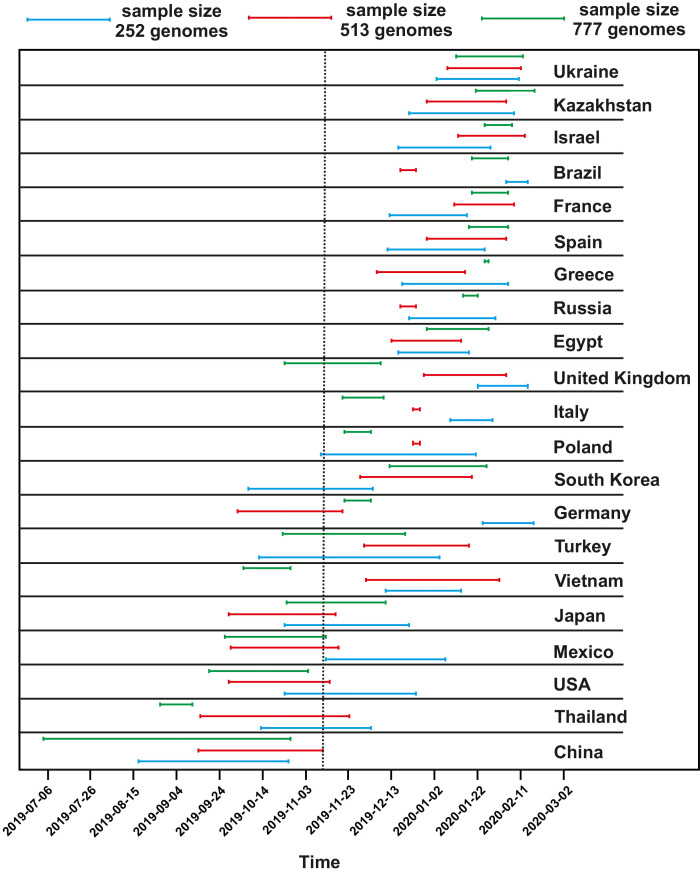
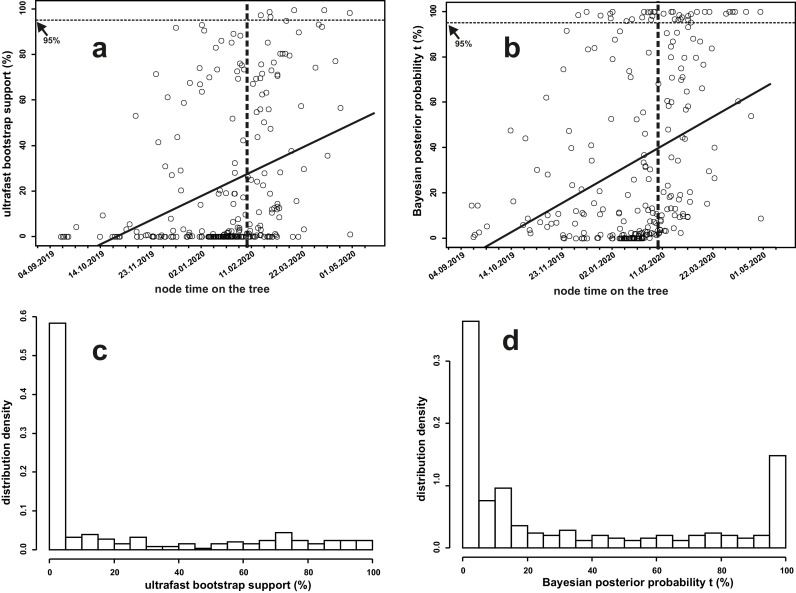
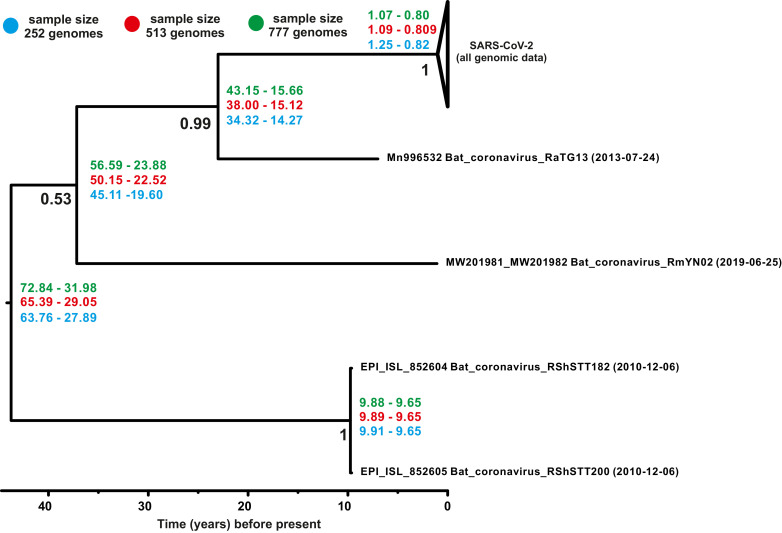
Samples from complete genomes of SARS-CoV-2 isolated during the first wave (December 2019-July 2020) of the global COVID-19 pandemic from 21 countries (Asia, Europe, Middle East and America) around the world, were analyzed using the phylogenetic method with molecular clock dating. Results showed that the first cases of COVID-19 in the human population appeared in the period between July and November 2019 in China. The spread of the virus into other countries of the world began in the autumn of 2019. In mid-February 2020, the virus appeared in all the countries we analyzed. During this time, the global population of SARS-CoV-2 was characterized by low levels of the genetic polymorphism, making it difficult to accurately assess the pathways of infection. The rate of evolution of the coding region of the SARS-CoV-2 genome equal to 7.3 × 10-4 (5.95 × 10-4-8.68 × 10-4) nucleotide substitutions per site per year is comparable to those of other human RNA viruses (Measles morbillivirus, Rubella virus, Enterovirus C). SARS-CoV-2 was separated from its known close relative, the bat coronavirus RaTG13 of the genus Betacoronavirus, approximately 15-43 years ago (the end of the 20th century).
Keywords: COVID-19; Evolutionary rate; Genomics; Molecular clock; SARS-CoV-2; Virus spread.
Copyright © 2021 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
