

A structural-based machine learning method to classify binding affinities between TCR and peptide-MHC complexes
- PMID: 34455212
- PMCID: PMC10811653
- DOI: 10.1016/j.molimm.2021.07.020
A structural-based machine learning method to classify binding affinities between TCR and peptide-MHC complexes
Abstract
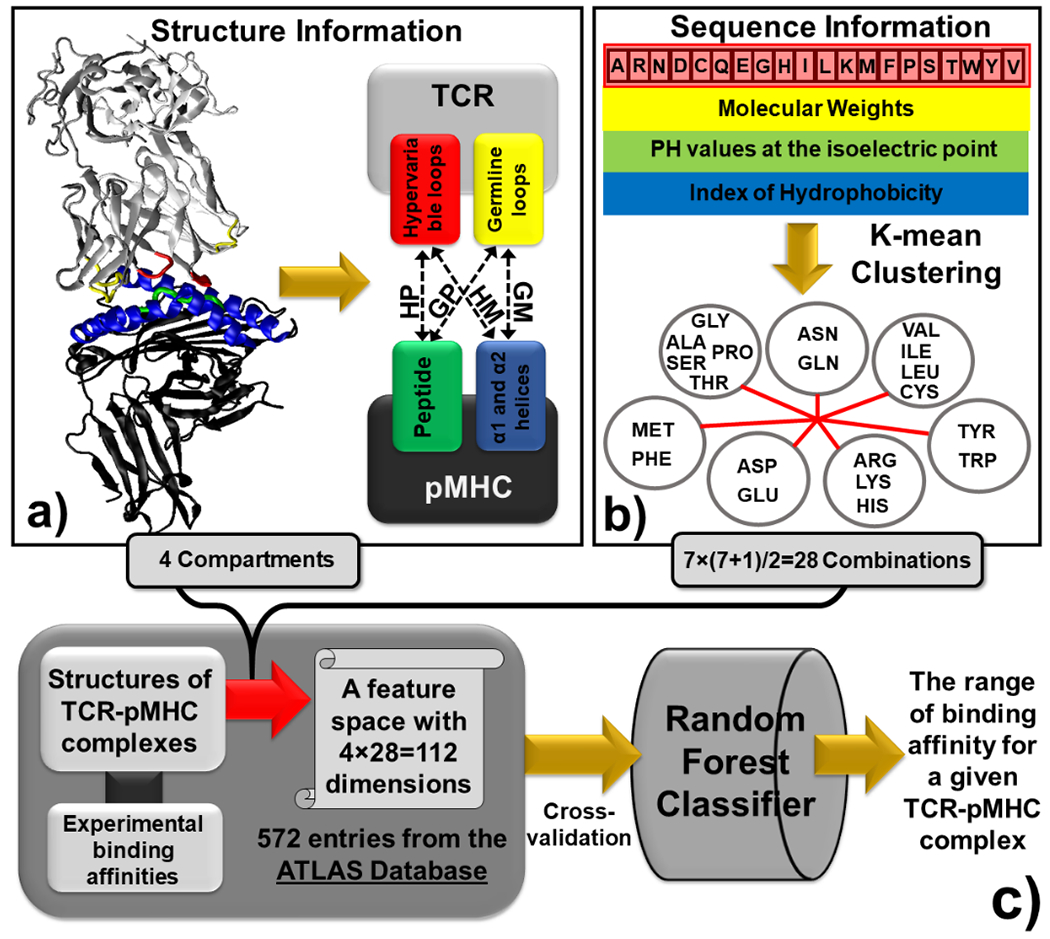
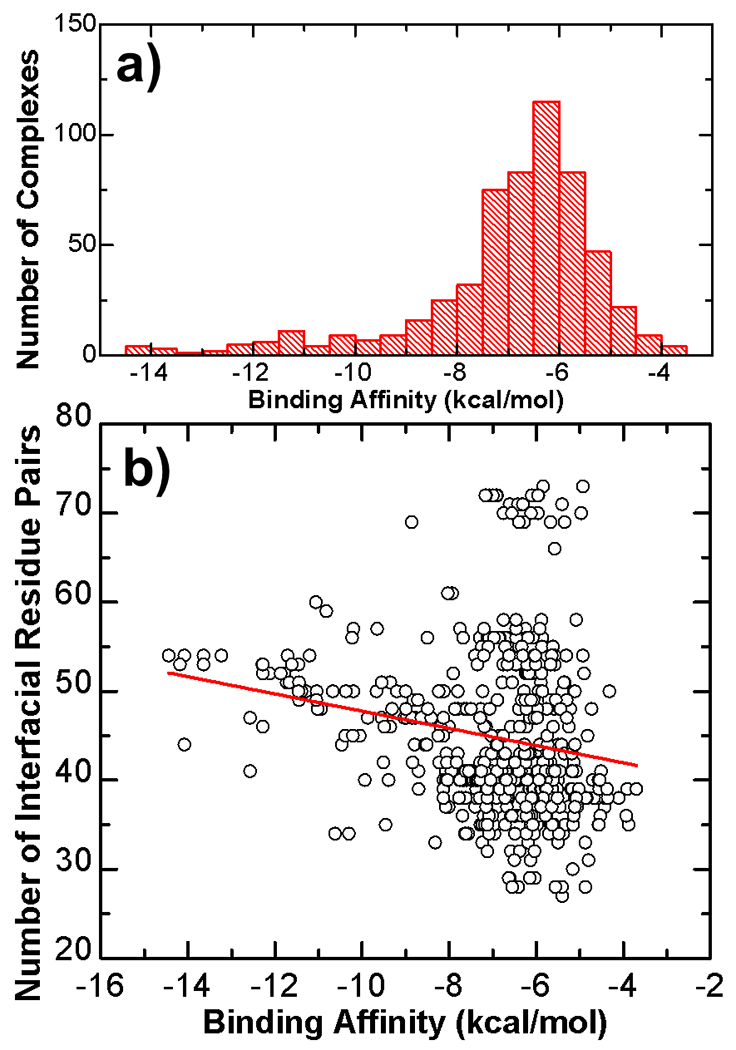
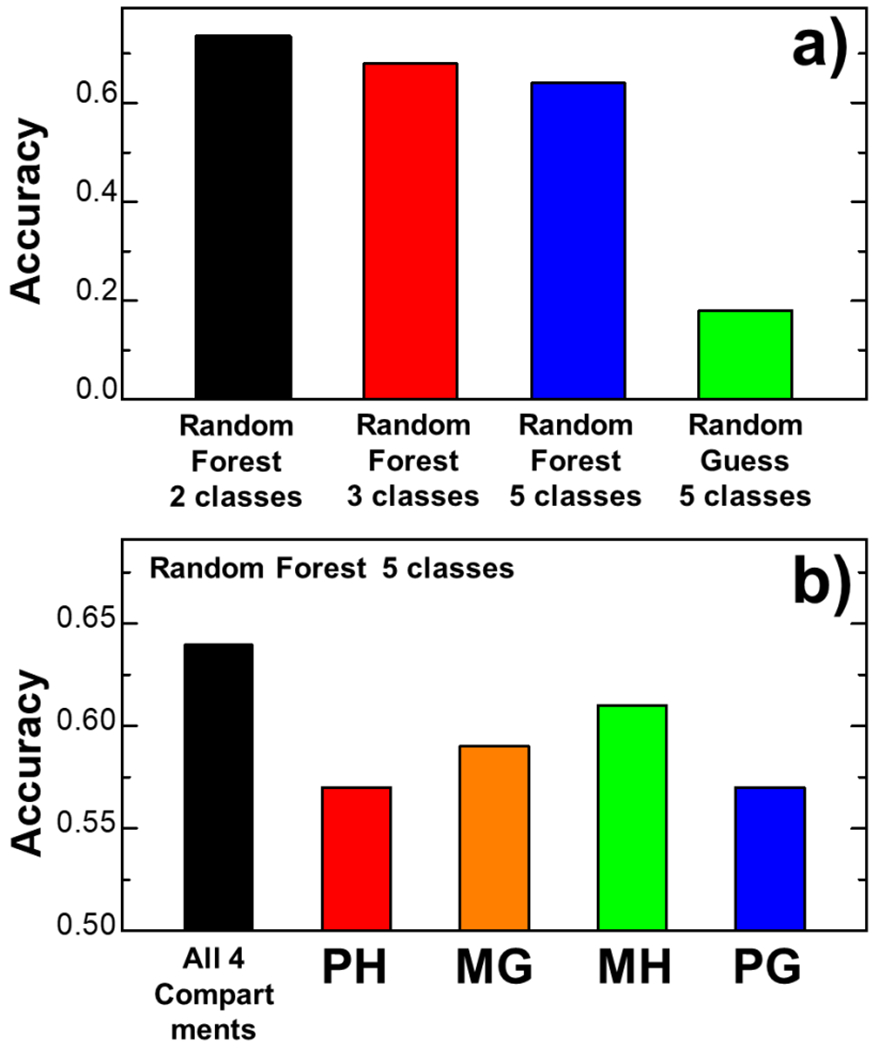
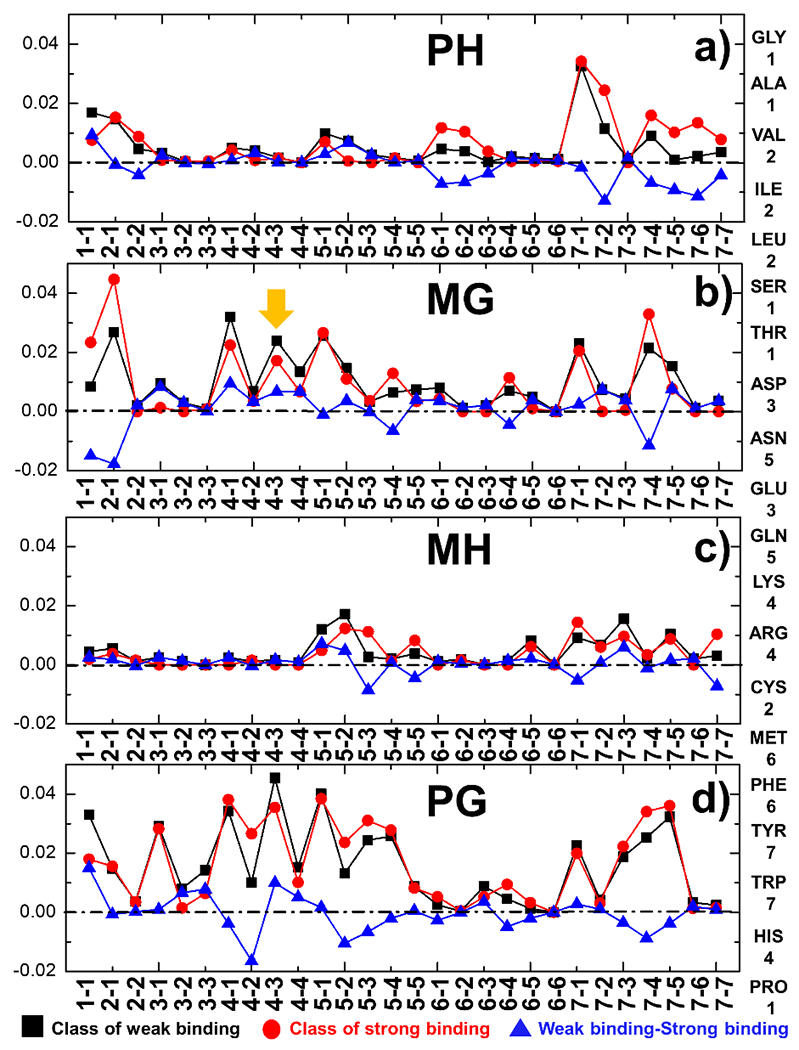
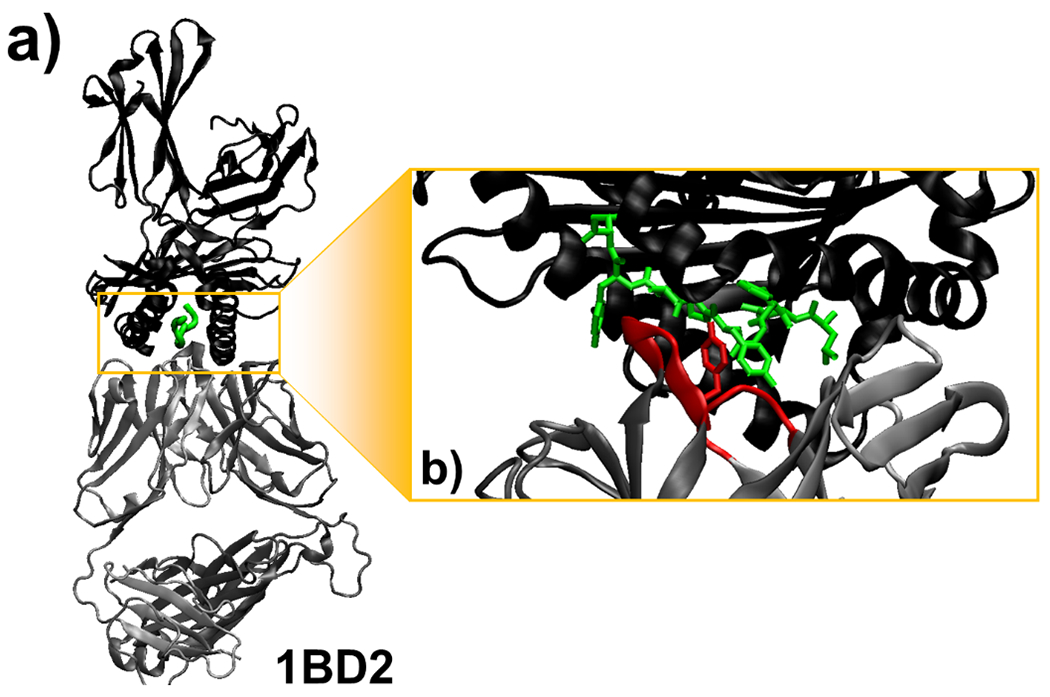
The activation of T cells is triggered by the interactions of T cell receptors (TCRs) with their epitopes, which are peptides presented by major histocompatibility complex (MHC) on the surfaces of antigen presenting cells (APC). While each TCR can only recognize a specific subset from a large repertoire of peptide-MHC (pMHC) complexes, it is very often that peptides in this subset share little sequence similarity. This is known as the specificity and cross-reactivity of T cells, respectively. The binding affinities between different types of TCRs and pMHC are the major driving force to shape this specificity and cross-reactivity in T cell recognition. The binding affinities, furthermore, are determined by the sequence and structural properties at the interfaces between TCRs and pMHC. Fortunately, a wealth of data on binding and structures of TCR-pMHC interactions becomes publicly accessible in online resources, which offers us the opportunity to develop a random forest classifier for predicting the binding affinities between TCR and pMHC based on the structure of their complexes. Specifically, the structure and sequence of a given complex were projected onto a high-dimensional feature space as the input of the classifier, which was then trained by a large-scale benchmark dataset. Based on the cross-validation results, we found that our machine learning model can predict if the binding affinity of a given TCR-pMHC complex is stronger or weaker than a predefined threshold with an overall accuracy approximately around 75 %. The significance of our prediction was estimated by statistical analysis. Moreover, more than 60 % of binding affinities in the ATLAS database can be successfully classified into groups within the range of 2 kcal/mol. Additionally, we show that TCR-pMHC complexes with strong binding affinity prefer hydrophobic interactions between amino acids with large aromatic rings instead of electrostatic interactions. Our results therefore provide insights to design engineered TCRs which enhance the specificity for their targeted epitopes. Taken together, this method can serve as a useful addition to a suite of existing approaches which study binding between TCR and pMHC.
Keywords: Binding affinity; Random forest classifier; TCR-pMHC complexes.
Copyright © 2021 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
