

Genomic and Phenotypic Biology of Novel Strains of Dickeya zeae Isolated From Pineapple and Taro in Hawaii: Insights Into Genome Plasticity, Pathogenicity, and Virulence Determinants
- PMID: 34456933
- PMCID: PMC8386352
- DOI: 10.3389/fpls.2021.663851
Genomic and Phenotypic Biology of Novel Strains of Dickeya zeae Isolated From Pineapple and Taro in Hawaii: Insights Into Genome Plasticity, Pathogenicity, and Virulence Determinants
Abstract
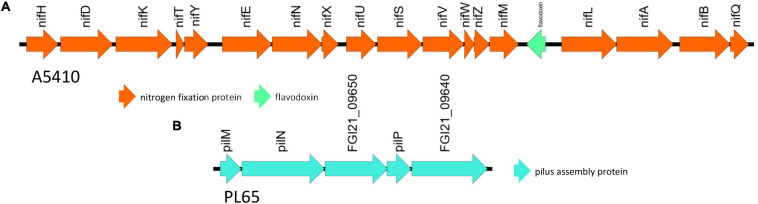
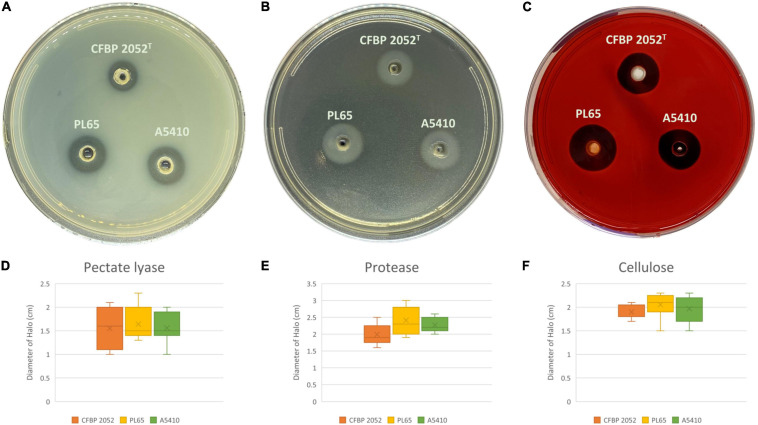
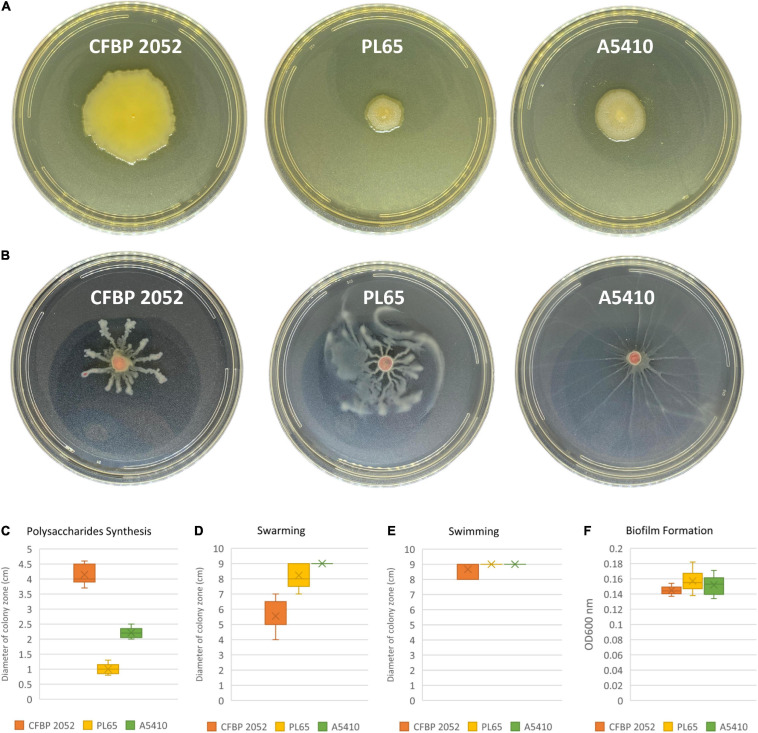
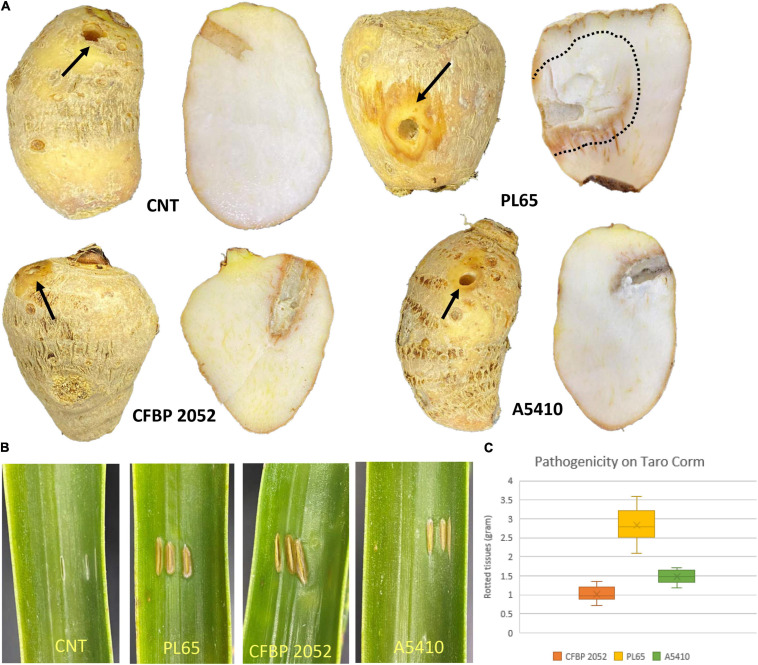
Dickeya zeae, a bacterial plant pathogen of the family Pectobacteriaceae, is responsible for a wide range of diseases on potato, maize, rice, banana, pineapple, taro, and ornamentals and significantly reduces crop production. D. zeae causes the soft rot of taro (Colocasia esculenta) and the heart rot of pineapple (Ananas comosus). In this study, we used Pacific Biosciences single-molecule real-time (SMRT) sequencing to sequence two high-quality complete genomes of novel strains of D. zeae: PL65 (size: 4.74997 MB; depth: 701x; GC: 53.6%) and A5410 (size: 4.7792 MB; depth: 558x; GC: 53.5%) isolated from economically important Hawaiian crops, taro, and pineapple, respectively. Additional complete genomes of D. zeae representing three additional hosts (philodendron, rice, and banana) and other species used for a taxonomic comparison were retrieved from the NCBI GenBank genome database. Genomic analyses indicated the truncated type III and IV secretion systems (T3SS and T4SS) in the taro strain, which only harbored one and two genes of T3SS and T4SS, respectively, and showed high heterogeneity in the type VI secretion system (T6SS). Unlike strain EC1, which was isolated from rice and recently reclassified as D. oryzae, neither the genome PL65 nor A5410 harbors the zeamine biosynthesis gene cluster, which plays a key role in virulence of other Dickeya species. The percentages of average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) between the two genomes were 94.47 and 57.00, respectively. In this study, we compared the major virulence factors [plant cell wall-degrading extracellular enzymes and protease (Prt)] produced by D. zeae strains and evaluated the virulence on taro corms and pineapple leaves. Both strains produced Prts, pectate lyases (Pels), and cellulases but no significant quantitative differences were observed (p > 0.05) between the strains. All the strains produced symptoms on taro corms and pineapple leaves, but the strain PL65 produced symptoms more rapidly than others. Our study highlights the genetic constituents of pathogenicity determinants and genomic heterogeneity that will help to understand the virulence mechanisms and aggressiveness of this plant pathogen.
Keywords: Dickeya zeae; comparative genomics; pathogenicity determinants and virulence factors; pectinolytic bacteria; phylogenomics; pineapple; taro (Colocasia esculenta).
Copyright © 2021 Boluk, Arizala, Dobhal, Zhang, Hu, Alvarez and Arif.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
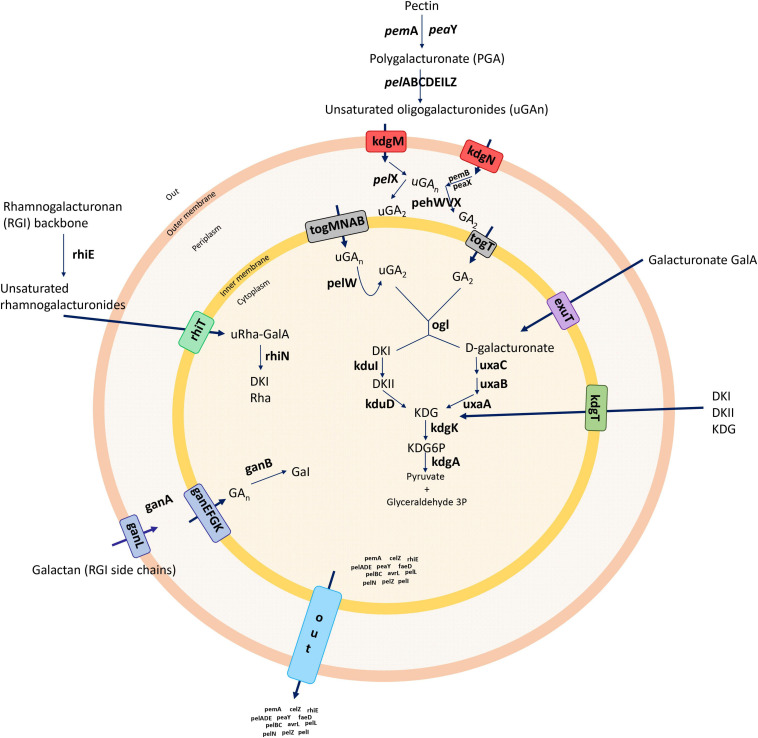
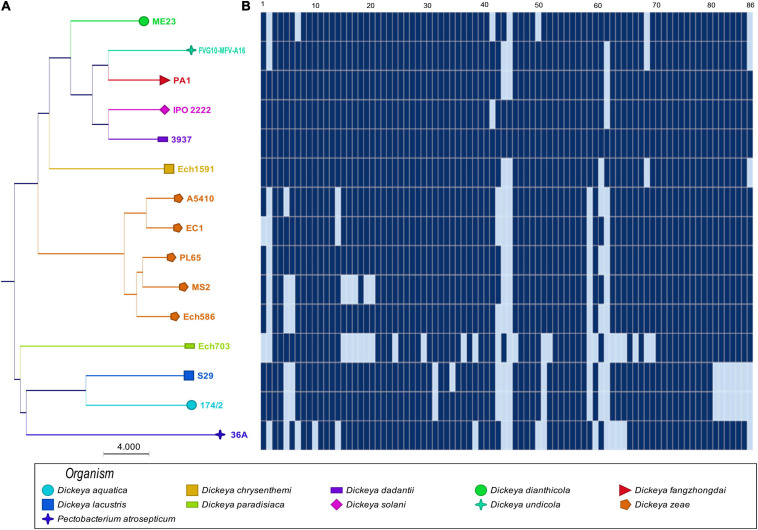
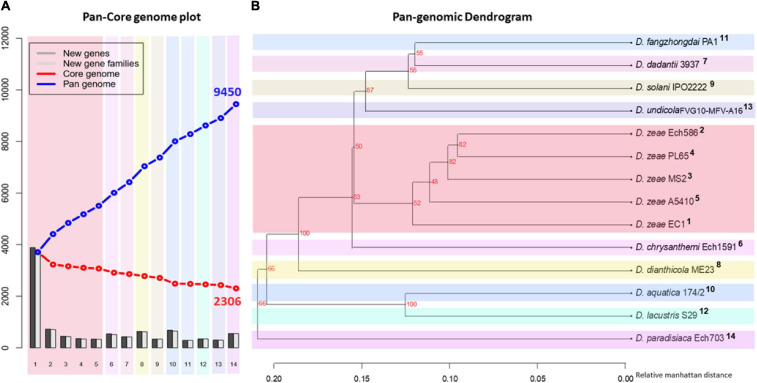
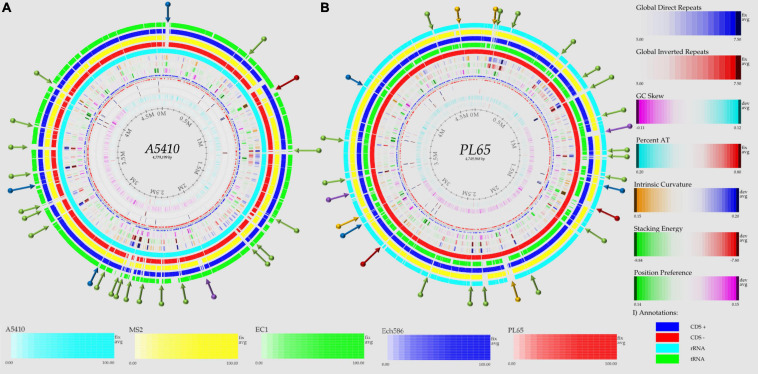
 ) shows the presence of genes, and light blue (
) shows the presence of genes, and light blue ( ) shows the absence of genes. In (B), numbers 1–86 are described in Supplementary Table 1.
) shows the absence of genes. In (B), numbers 1–86 are described in Supplementary Table 1.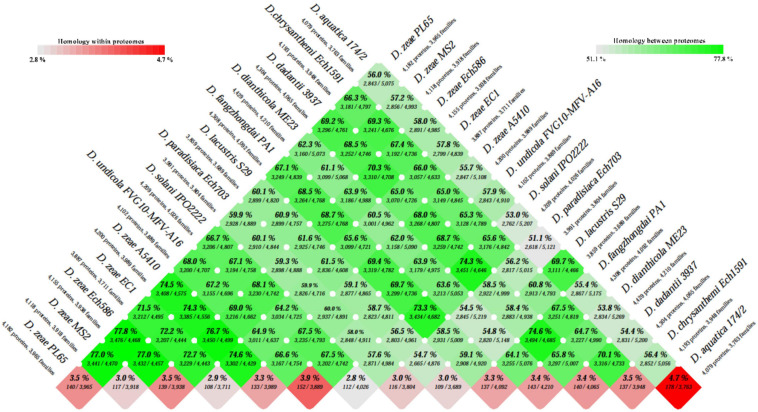
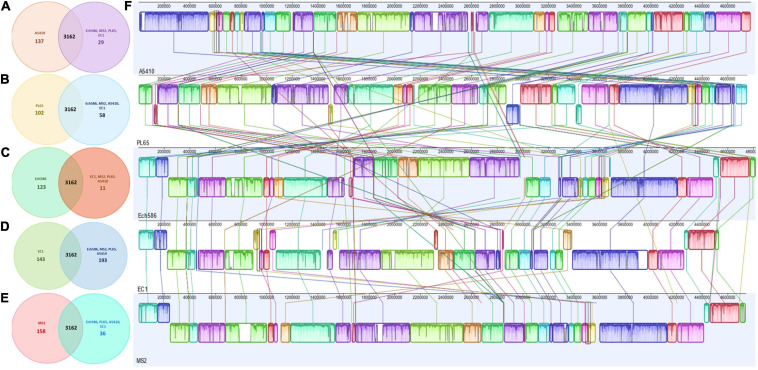
 ); the order of contigs (shown in
); the order of contigs (shown in  ); distribution of coding sequences in forward strands (shown in
); distribution of coding sequences in forward strands (shown in  ); distribution of coding sequences in reverse strands (shown in
); distribution of coding sequences in reverse strands (shown in  ); distribution of non-coding elements along the chromosome (shown in
); distribution of non-coding elements along the chromosome (shown in  ); distribution of genes involved in antibiotic resistance (shown in
); distribution of genes involved in antibiotic resistance (shown in  ); distribution of other virulence factors (shown in
); distribution of other virulence factors (shown in  ); distribution of genes encoding transporter proteins (shown in
); distribution of genes encoding transporter proteins (shown in  ); distribution of genes encoding drug targets (shown in
); distribution of genes encoding drug targets (shown in  ); distribution of GC content (shown in
); distribution of GC content (shown in  ); and distribution of GC skew (shown in
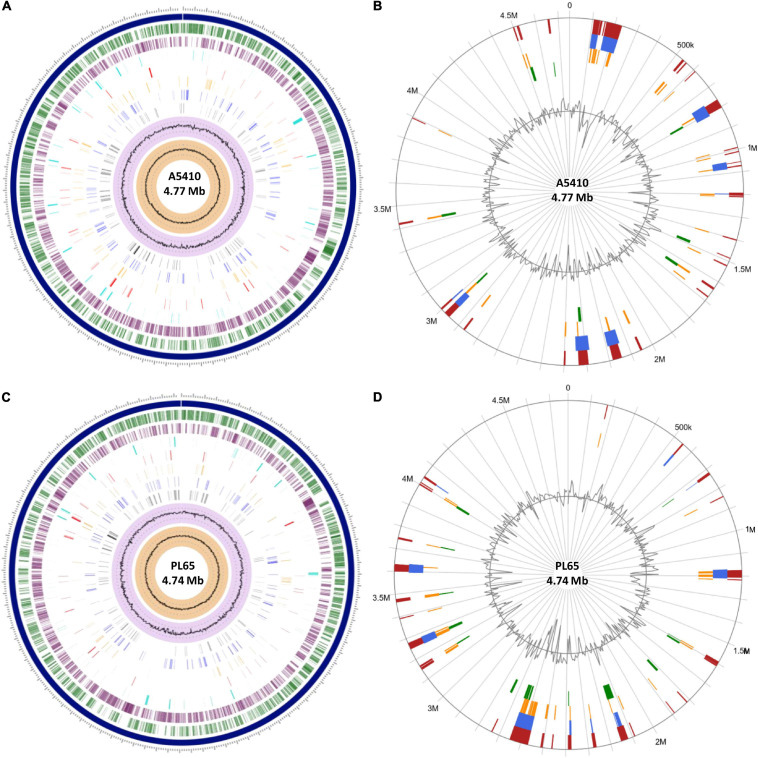
); and distribution of GC skew (shown in  ). Circular visualization of the predicted Genomic Islands (GIs) on A5410 (B) and PL65 (D) strains. The analysis was conducted in IslandViewer 4. The interactive visualization of the distinct islands across the genomes is shown with blocks colored according to the predictor tool as described: IslandPick (shown in
). Circular visualization of the predicted Genomic Islands (GIs) on A5410 (B) and PL65 (D) strains. The analysis was conducted in IslandViewer 4. The interactive visualization of the distinct islands across the genomes is shown with blocks colored according to the predictor tool as described: IslandPick (shown in  ) based on genome comparison, IslandPath-DIMOB (shown in
) based on genome comparison, IslandPath-DIMOB (shown in  ) based on associated GIs features such as transfer RNAs (tRNAs), transposon elements, integrases, and sequence bias, SIGI-HMM (shown in
) based on associated GIs features such as transfer RNAs (tRNAs), transposon elements, integrases, and sequence bias, SIGI-HMM (shown in  ), based on the codon usage bias with a Hidden Markov model criterion and the integrated results of the four tools (shown in
), based on the codon usage bias with a Hidden Markov model criterion and the integrated results of the four tools (shown in  ).
).
References
-
- Adeolu M., Alnajar S., Naushad S., Gupta R. S. (2016). Genome-based phylogeny, and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacteriales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 66 5575–5599. 10.1099/ijsem.0.001485 - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous