

Recent Developments in Free Energy Calculations for Drug Discovery
- PMID: 34458321
- PMCID: PMC8387144
- DOI: 10.3389/fmolb.2021.712085
Recent Developments in Free Energy Calculations for Drug Discovery
Abstract
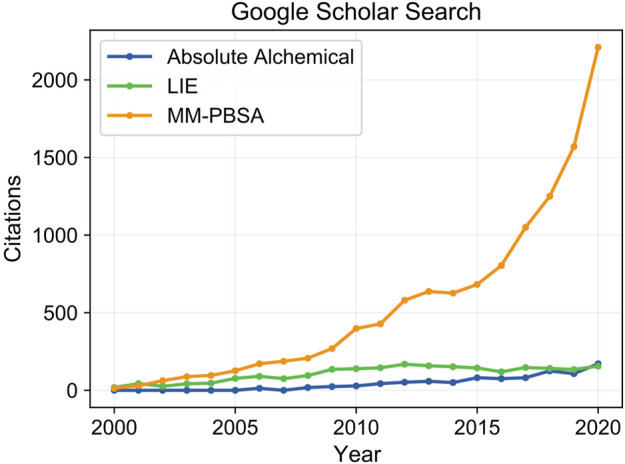
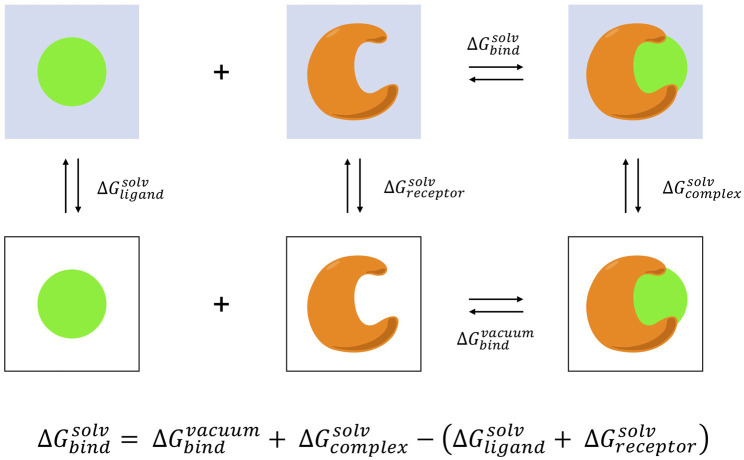
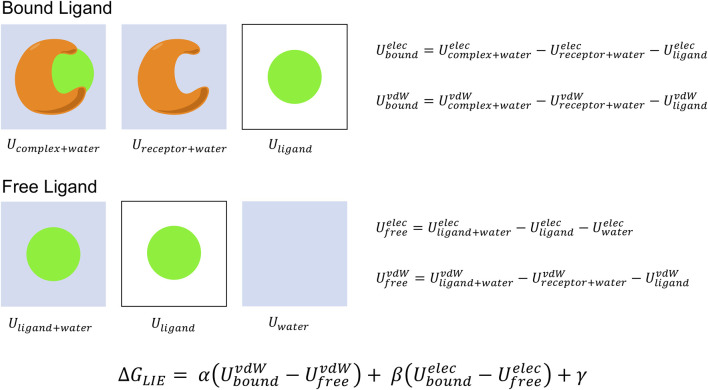
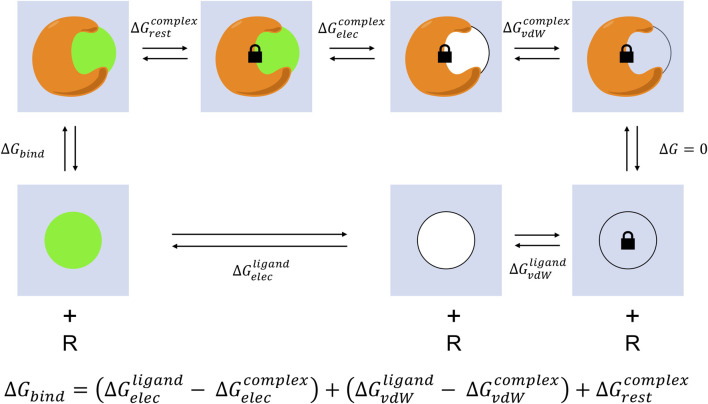
The grand challenge in structure-based drug design is achieving accurate prediction of binding free energies. Molecular dynamics (MD) simulations enable modeling of conformational changes critical to the binding process, leading to calculation of thermodynamic quantities involved in estimation of binding affinities. With recent advancements in computing capability and predictive accuracy, MD based virtual screening has progressed from the domain of theoretical attempts to real application in drug development. Approaches including the Molecular Mechanics Poisson Boltzmann Surface Area (MM-PBSA), Linear Interaction Energy (LIE), and alchemical methods have been broadly applied to model molecular recognition for drug discovery and lead optimization. Here we review the varied methodology of these approaches, developments enhancing simulation efficiency and reliability, remaining challenges hindering predictive performance, and applications to problems in the fields of medicine and biochemistry.
Keywords: MM-PBSA; alchemical simulation; binding affinity; drug discovery; free energy simulation; lie; molecular dynamics.
Copyright © 2021 King, Aitchison, Li and Luo.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Calculate protein-ligand binding affinities with the extended linear interaction energy method: application on the Cathepsin S set in the D3R Grand Challenge 3.J Comput Aided Mol Des. 2019 Jan;33(1):105-117. doi: 10.1007/s10822-018-0162-6. Epub 2018 Sep 14. J Comput Aided Mol Des. 2019. PMID: 30218199 Free PMC article.
-
Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations.J Chem Inf Model. 2017 Dec 26;57(12):2911-2937. doi: 10.1021/acs.jcim.7b00564. Epub 2017 Dec 15. J Chem Inf Model. 2017. PMID: 29243483
-
How Well Does the Extended Linear Interaction Energy Method Perform in Accurate Binding Free Energy Calculations?J Chem Inf Model. 2020 Dec 28;60(12):6624-6633. doi: 10.1021/acs.jcim.0c00934. Epub 2020 Nov 19. J Chem Inf Model. 2020. PMID: 33213150
-
Free energy calculations to estimate ligand-binding affinities in structure-based drug design.Curr Pharm Des. 2014;20(20):3323-37. doi: 10.2174/13816128113199990604. Curr Pharm Des. 2014. PMID: 23947646 Review.
-
The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities.Expert Opin Drug Discov. 2015 May;10(5):449-61. doi: 10.1517/17460441.2015.1032936. Epub 2015 Apr 2. Expert Opin Drug Discov. 2015. PMID: 25835573 Free PMC article. Review.
Cited by
-
Structural bioinformatics for rational drug design.Res Pract Thromb Haemost. 2025 Jan 23;9(1):102691. doi: 10.1016/j.rpth.2025.102691. eCollection 2025 Jan. Res Pract Thromb Haemost. 2025. PMID: 40027444 Free PMC article.
-
Substituents introduction of methyl and methoxy functional groups on resveratrol stabilizes mTOR binding for autophagic cell death induction.Sci Rep. 2025 Apr 26;15(1):14675. doi: 10.1038/s41598-025-98616-6. Sci Rep. 2025. PMID: 40287470 Free PMC article.
-
In silico approaches to identify novel anti-diabetic type 2 agents against dipeptidyl peptidase IV from isoxazole derivatives of usnic acid.3 Biotech. 2025 May;15(5):107. doi: 10.1007/s13205-025-04287-5. Epub 2025 Apr 2. 3 Biotech. 2025. PMID: 40191458
-
Binding free energies for the SAMPL8 CB8 "Drugs of Abuse" challenge from umbrella sampling combined with Hamiltonian replica exchange.J Comput Aided Mol Des. 2022 Jan;36(1):1-9. doi: 10.1007/s10822-021-00439-w. Epub 2022 Jan 3. J Comput Aided Mol Des. 2022. PMID: 34978001 Free PMC article.
-
IMERGE-FEP: Improving Relative Free Energy Calculation Convergence with Chemical Intermediates.J Phys Chem B. 2025 Mar 6;129(9):2370-2379. doi: 10.1021/acs.jpcb.4c07156. Epub 2025 Feb 20. J Phys Chem B. 2025. PMID: 39976528 Free PMC article.
References
-
- Al-Madhagi W. M., Hashim N. M., Awadh Ali N. A., Taha H., Alhadi A. A., Abdullah A. A., et al. (2019). Bioassay-Guided Isolation and In Silico Study of Antibacterial Compounds from Petroleum Ether Extract of Peperomia Blanda (Jacq.) Kunth. J. Chem. Inf. Model. 59, 1858–1872. 10.1021/acs.jcim.8b00969 - DOI - PubMed
-
- Aldeghi M., Bodkin M. J., Knapp S., Biggin P. C. (2017). Statistical Analysis on the Performance of Molecular Mechanics Poisson-Boltzmann Surface Area versus Absolute Binding Free Energy Calculations: Bromodomains as a Case Study. J. Chem. Inf. Model. 57, 2203–2221. 10.1021/acs.jcim.7b00347 - DOI - PMC - PubMed