

ω-Functionalized Lipid Prodrugs of HIV NtRTI Tenofovir with Enhanced Pharmacokinetic Properties
- PMID: 34459598
- PMCID: PMC12010929
- DOI: 10.1021/acs.jmedchem.1c01083
ω-Functionalized Lipid Prodrugs of HIV NtRTI Tenofovir with Enhanced Pharmacokinetic Properties
Abstract
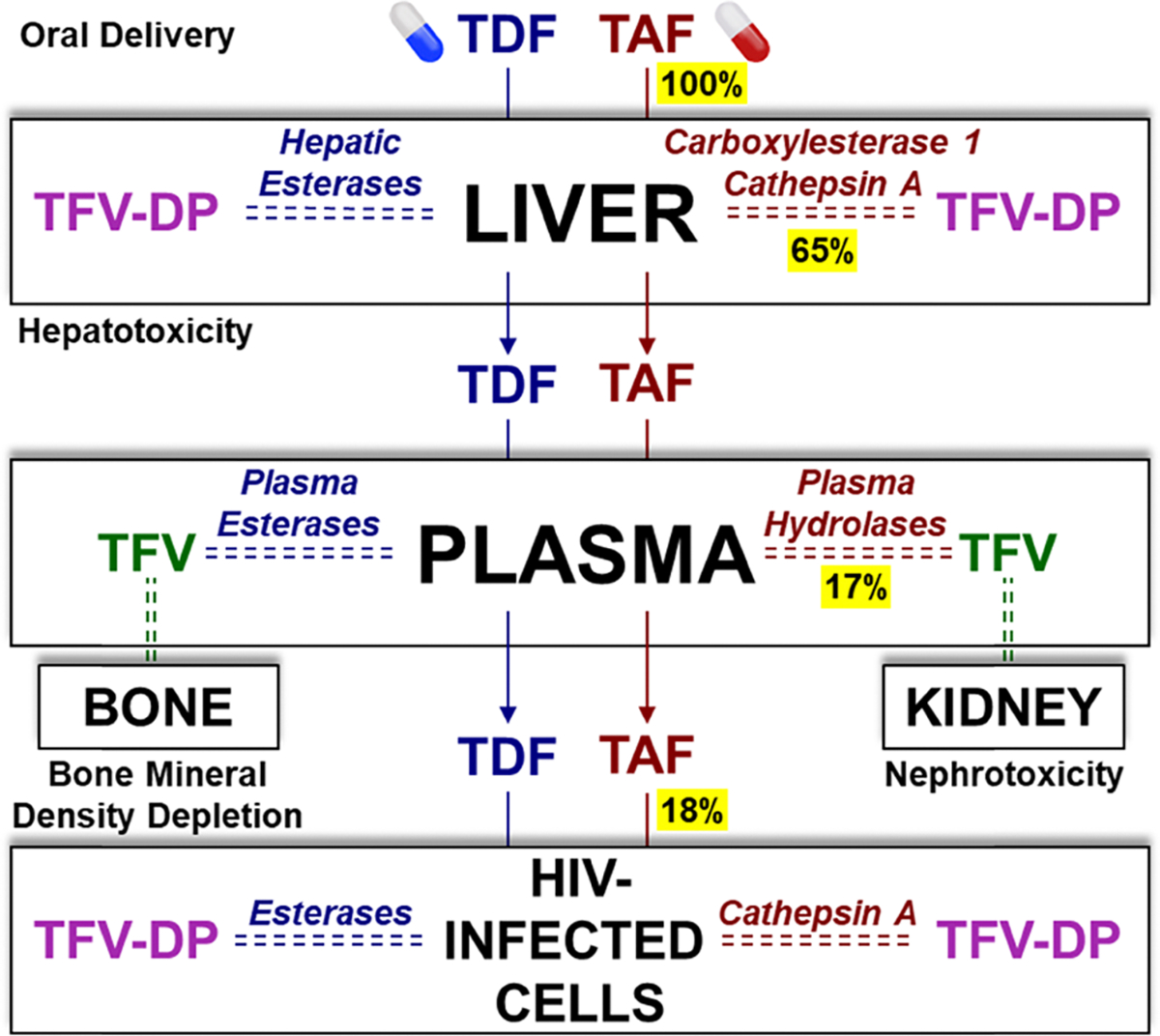
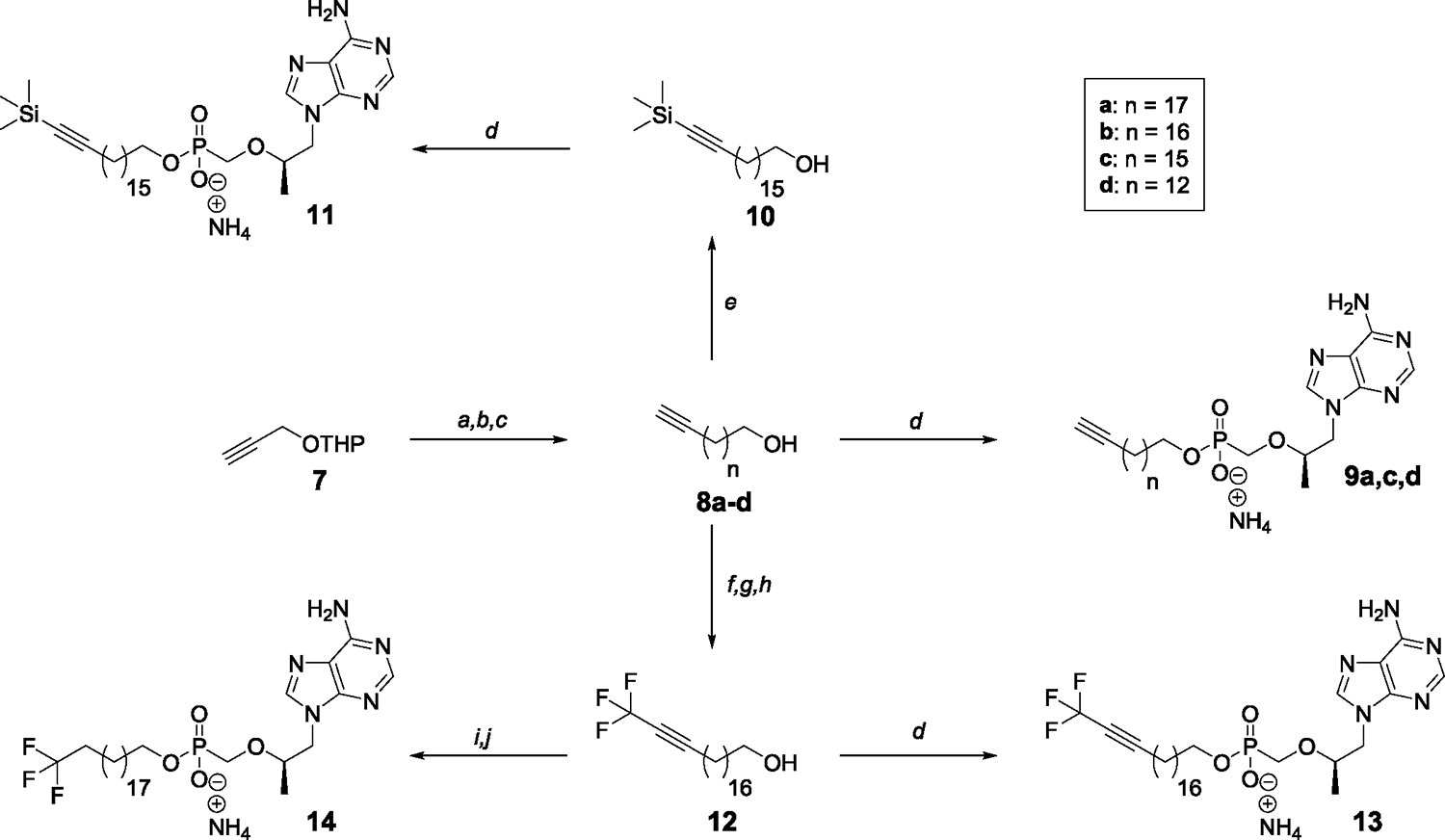
Tenofovir (TFV) is the cornerstone nucleotide reverse transcriptase inhibitor (NtRTI) in many combination antiretroviral therapies prescribed to patients living with HIV/AIDS. Due to poor cell permeability and oral bioavailability, TFV is administered as one of two FDA-approved prodrugs, both of which metabolize prematurely in the liver and/or plasma. This premature prodrug processing depletes significant fractions of each oral dose and causes toxicity in kidney, bone, and liver with chronic administration. Although TFV exalidex (TXL), a phospholipid-derived prodrug of TFV, was designed to address this issue, clinical pharmacokinetic studies indicated substantial hepatic extraction, redirecting clinical development of TXL toward HBV. To circumvent this metabolic liability, we synthesized and evaluated ω-functionalized TXL analogues with dramatically improved hepatic stability. This effort led to the identification of compounds 21 and 23, which exhibited substantially longer t1/2 values than TXL in human liver microsomes, potent anti-HIV activity in vitro, and enhanced pharmacokinetic properties in vivo.
Conflict of interest statement
The authors declare no competing financial interest.
Figures








References
-
- Brown NA Progress Towards Improving Antiviral Therapy for Hepatitis C with Hepatitis C Virus Polymerase Inhibitors. Part I: Nucleoside Analogues. Expert Opin. Investig. Drugs 2009, 18, 709–725. - PubMed
-
- Stein DS; Moore KHP Phosphorylation of Nucleoside Analog Antiretrovirals: A Review for Clinicians. Pharmacotherapy 2001, 21, 11–34. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Chemical Information
