

Conifer: clonal tree inference for tumor heterogeneity with single-cell and bulk sequencing data
- PMID: 34461827
- PMCID: PMC8404257
- DOI: 10.1186/s12859-021-04338-7
Conifer: clonal tree inference for tumor heterogeneity with single-cell and bulk sequencing data
Abstract
Background: Genetic heterogeneity of a cancer tumor that develops during clonal evolution is one of the reasons for cancer treatment failure, by increasing the chance of drug resistance. Clones are cell populations with different genotypes, resulting from differences in somatic mutations that occur and accumulate during cancer development. An appropriate approach for identifying clones is determining the variant allele frequency of mutations that occurred in the tumor. Although bulk sequencing data can be used to provide that information, the frequencies are not informative enough for identifying different clones with the same prevalence and their evolutionary relationships. On the other hand, single-cell sequencing data provides valuable information about branching events in the evolution of a cancerous tumor. However, the temporal order of mutations may be determined with ambiguities using only single-cell data, while variant allele frequencies from bulk sequencing data can provide beneficial information for inferring the temporal order of mutations with fewer ambiguities.
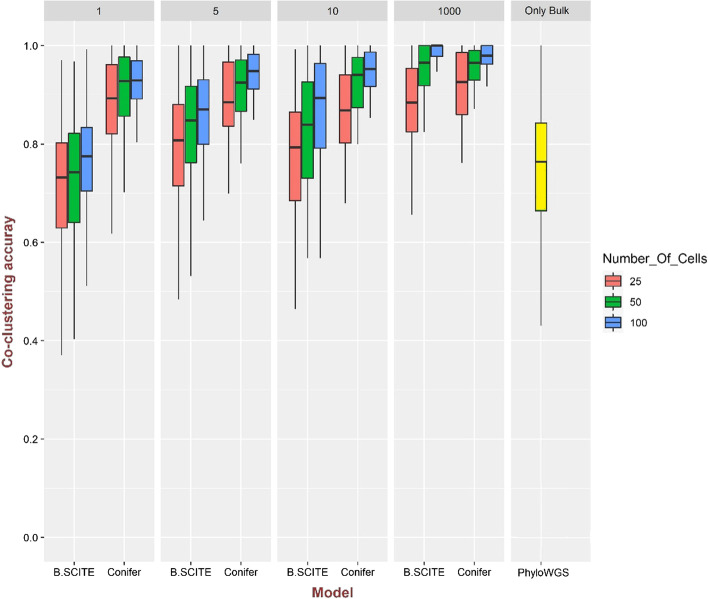
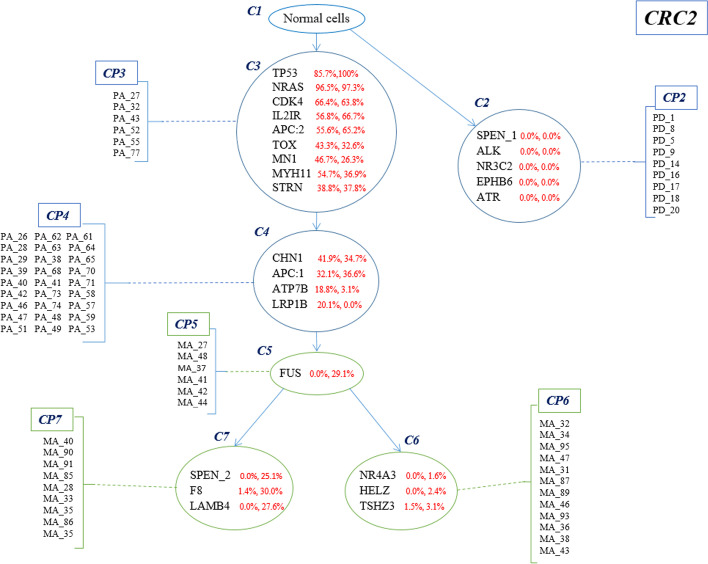
Result: In this study, a new method called Conifer (ClONal tree Inference For hEterogeneity of tumoR) is proposed which combines aggregated variant allele frequency from bulk sequencing data with branching event information from single-cell sequencing data to more accurately identify clones and their evolutionary relationships. It is proven that the accuracy of clone identification and clonal tree inference is increased by using Conifer compared to other existing methods on various sets of simulated data. In addition, it is discussed that the evolutionary tree provided by Conifer on real cancer data sets is highly consistent with information in both bulk and single-cell data.
Conclusions: In this study, we have provided an accurate and robust method to identify clones of tumor heterogeneity and their evolutionary history by combining single-cell and bulk sequencing data.
Keywords: Bayesian nonparametric model; Bulk sequencing; Clonal tree; Heterogeneity of tumor; Single-cell sequencing.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






References
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. - PubMed
-
- Marte B. Tumour heterogeneity. Berlin: Nature Publishing Group; 2013. - PubMed
-
- Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–334. - PubMed
-
- Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924–935. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
