

Microbial signatures in the lower airways of mechanically ventilated COVID-19 patients associated with poor clinical outcome
- PMID: 34465900
- PMCID: PMC8484067
- DOI: 10.1038/s41564-021-00961-5
Microbial signatures in the lower airways of mechanically ventilated COVID-19 patients associated with poor clinical outcome
Abstract
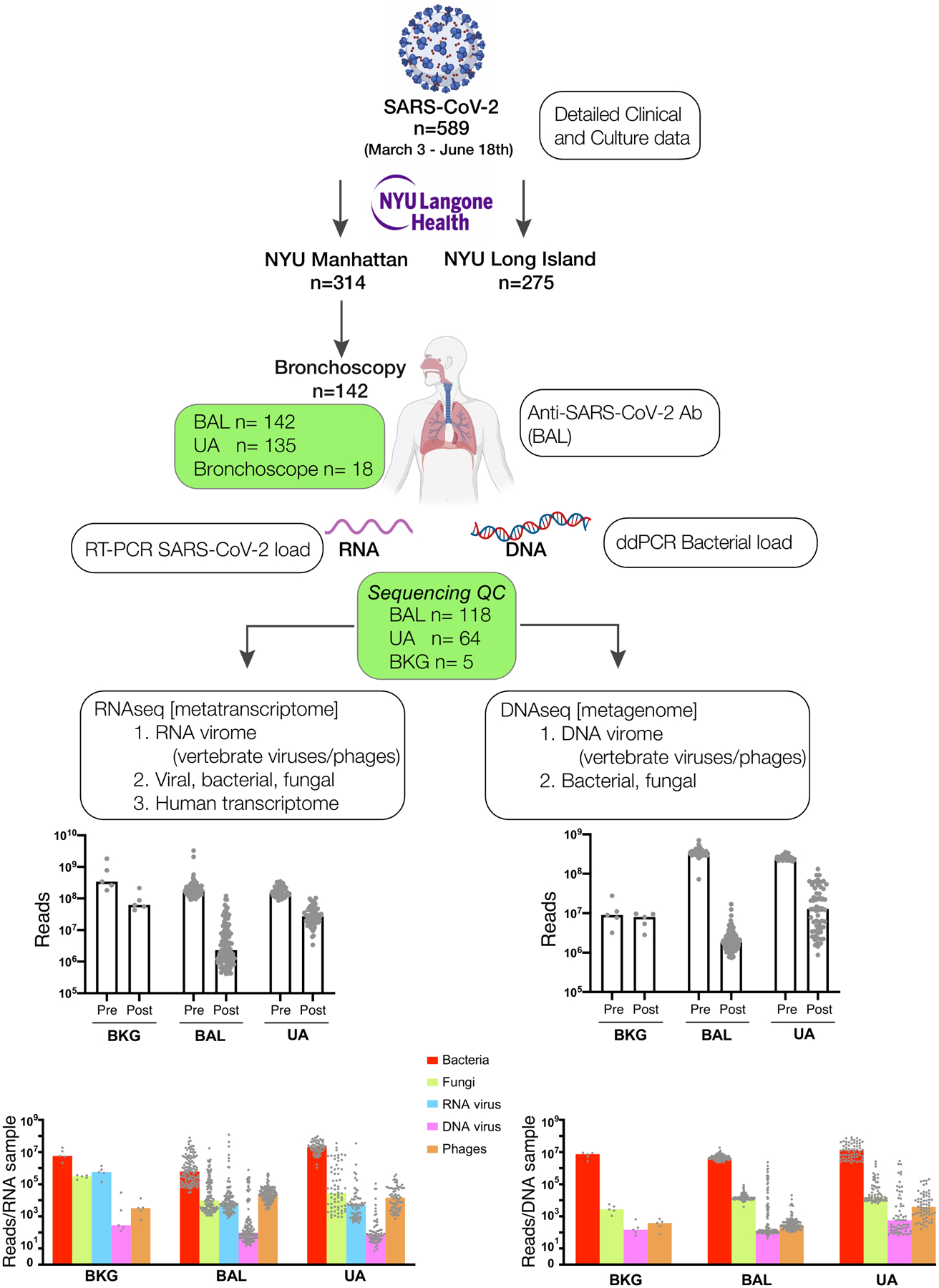
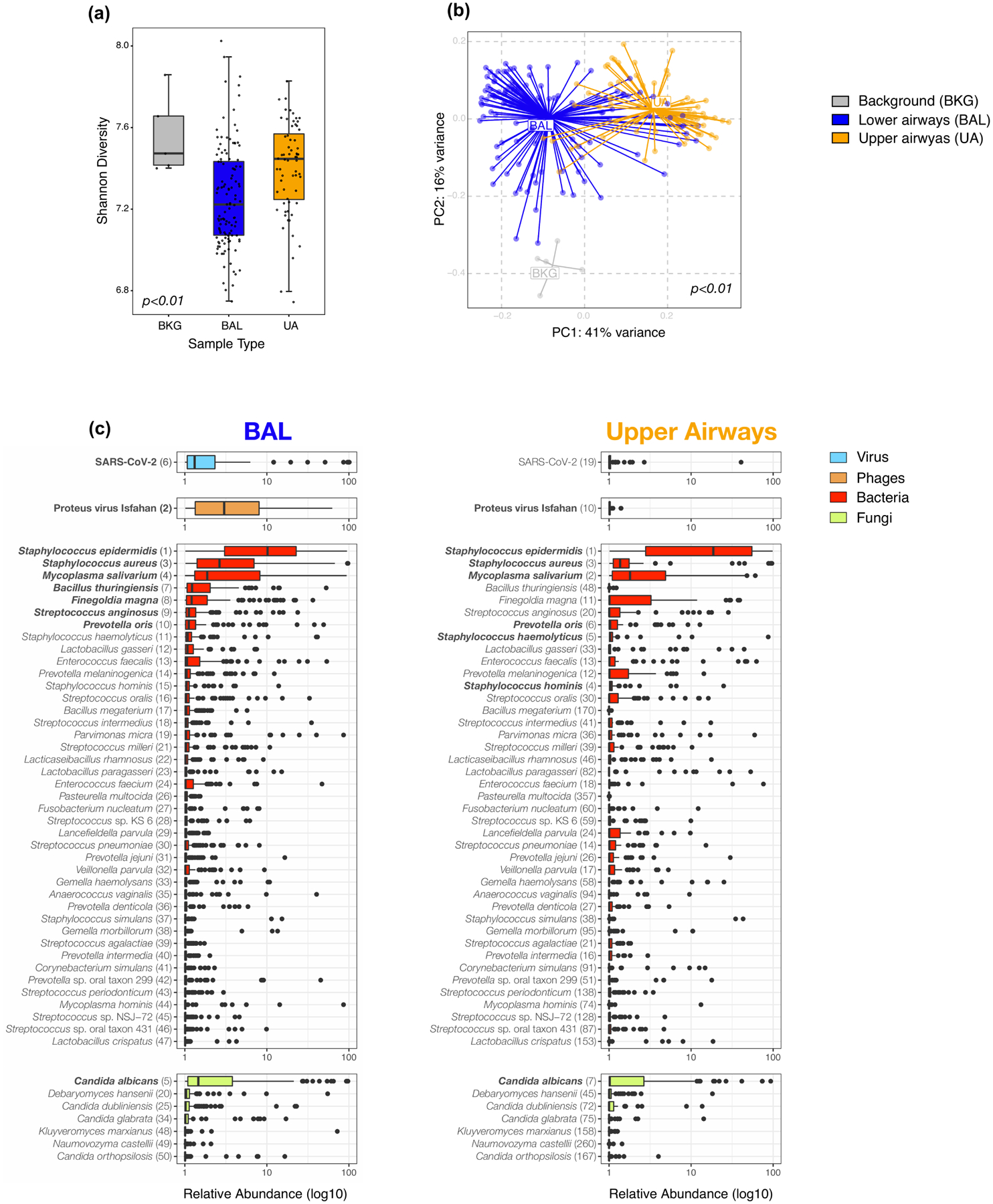
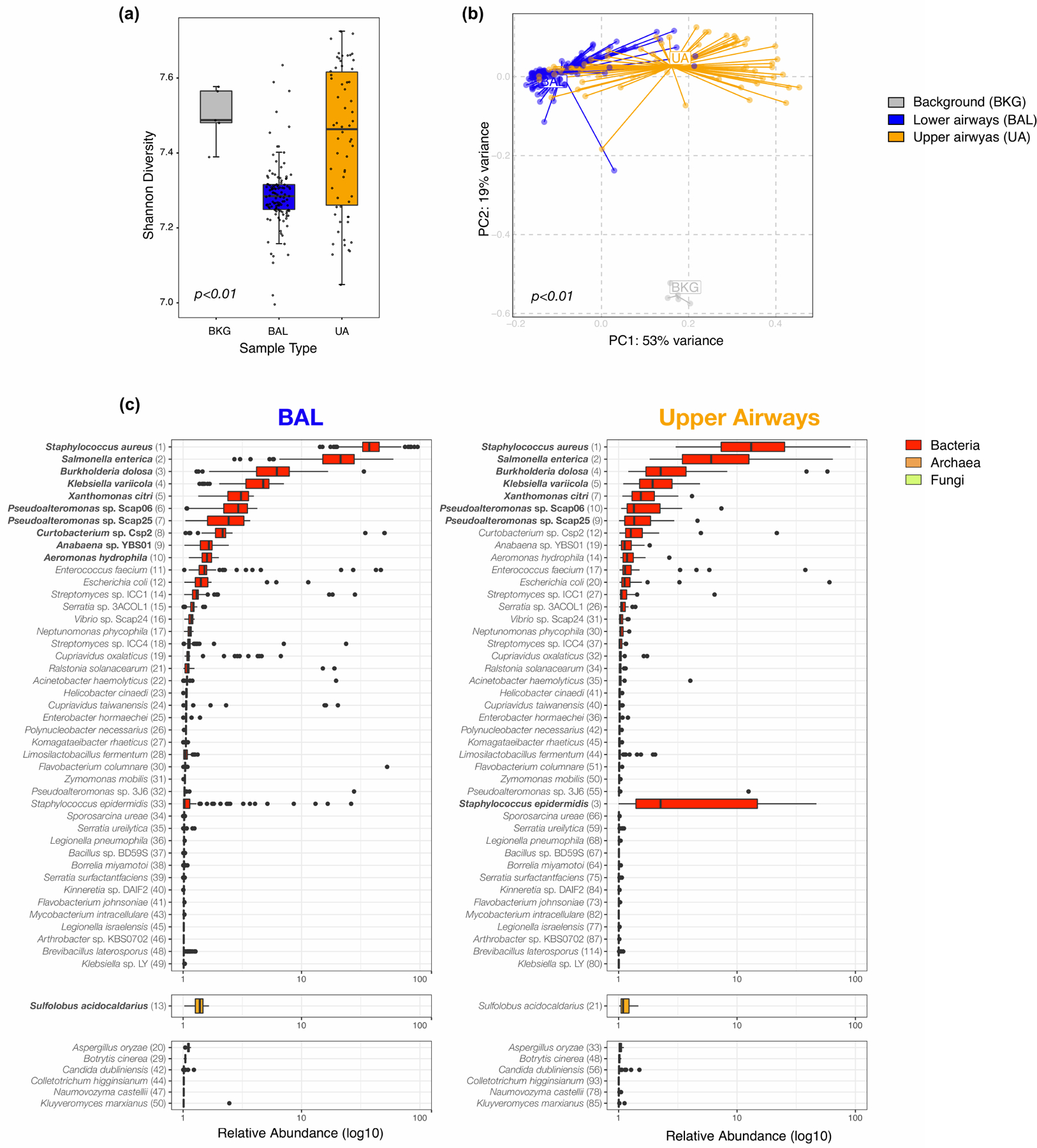
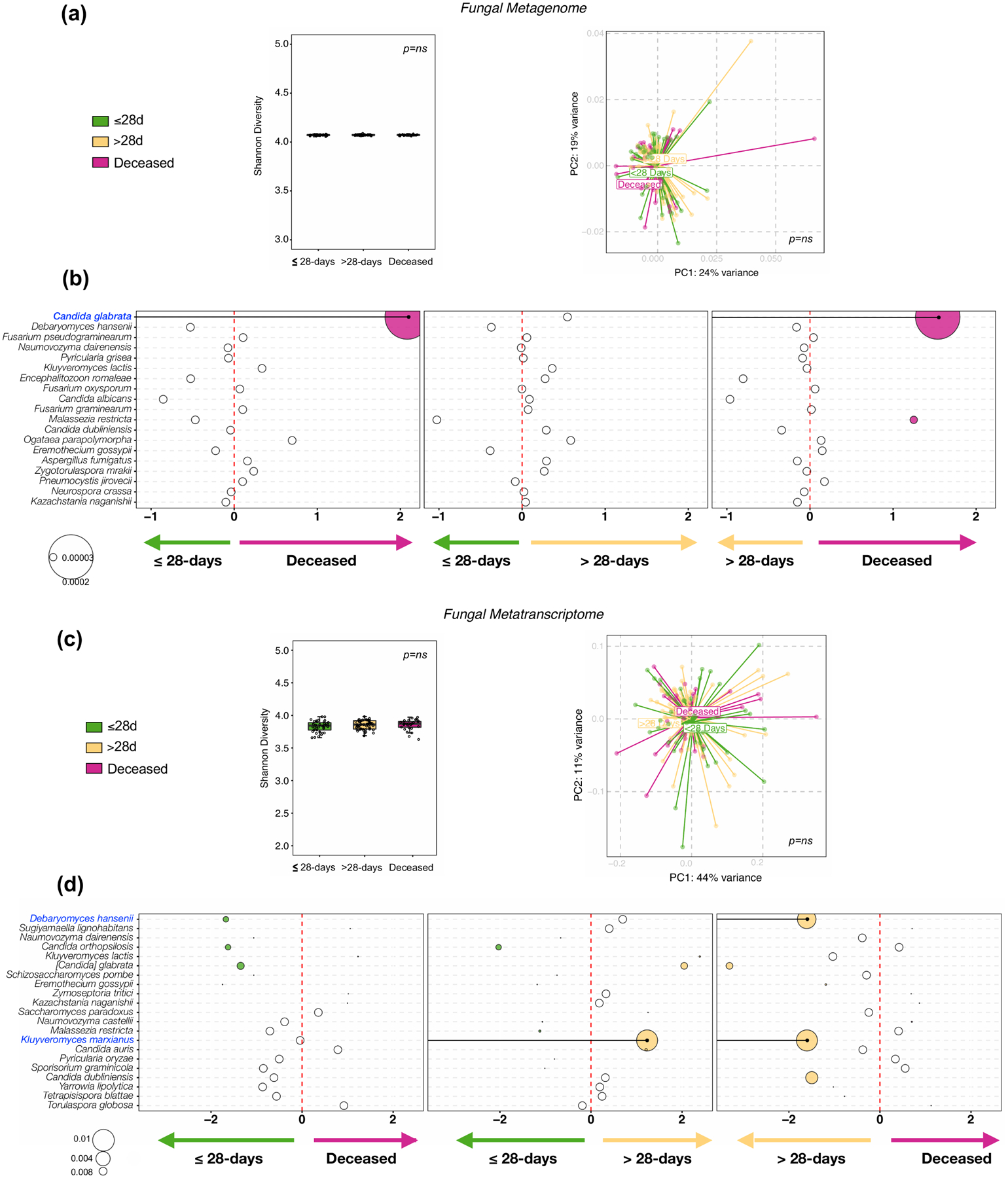
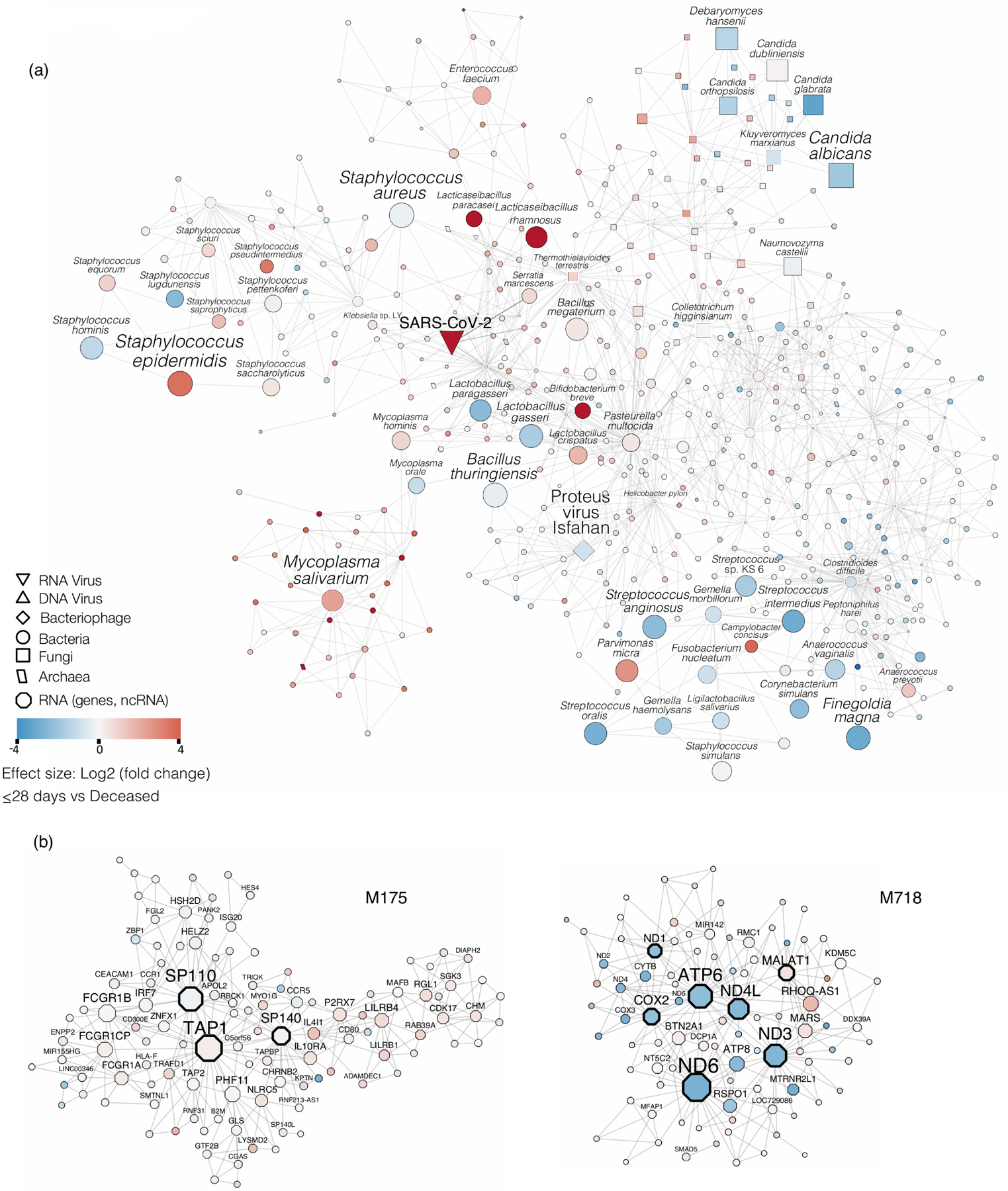
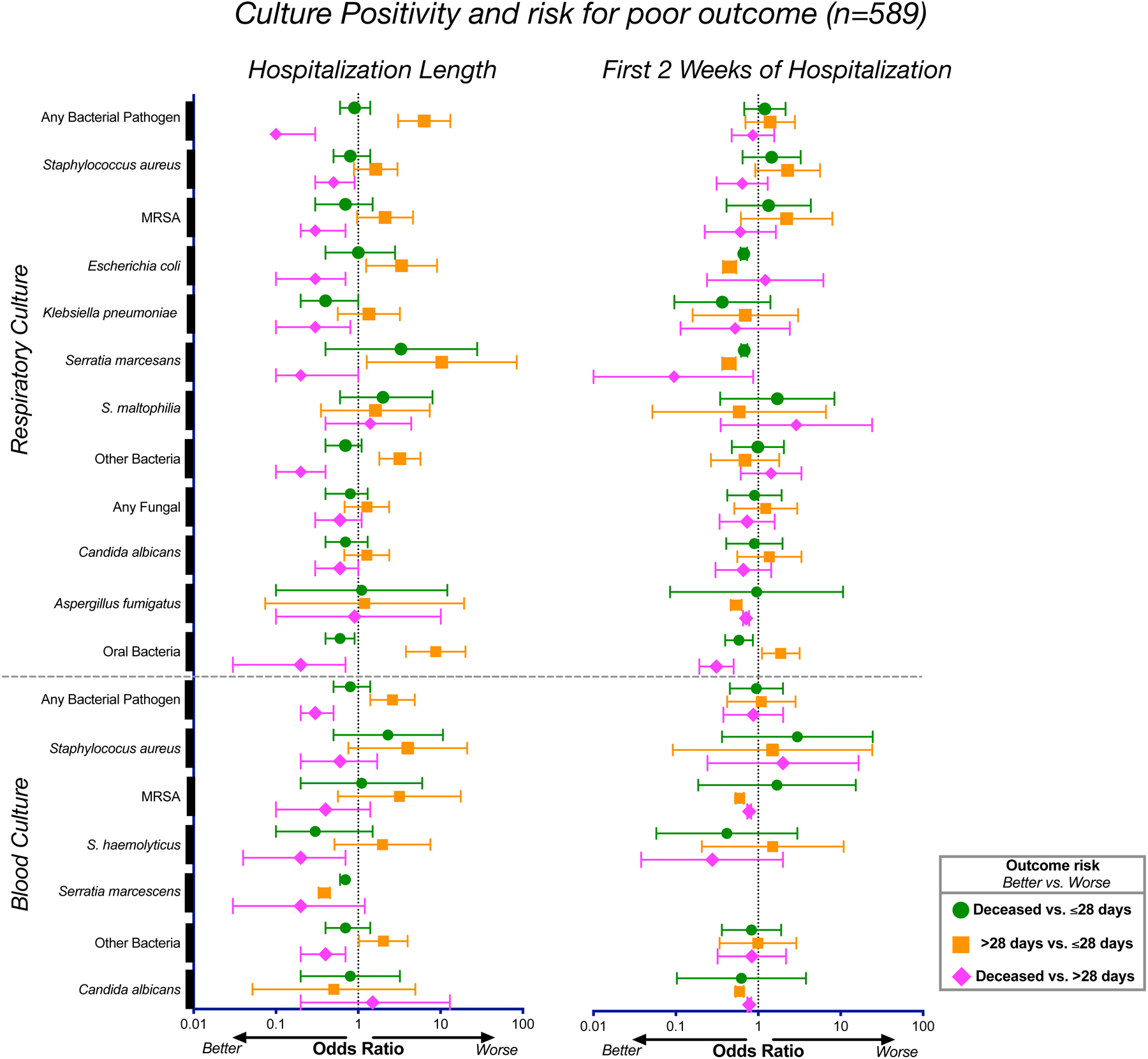
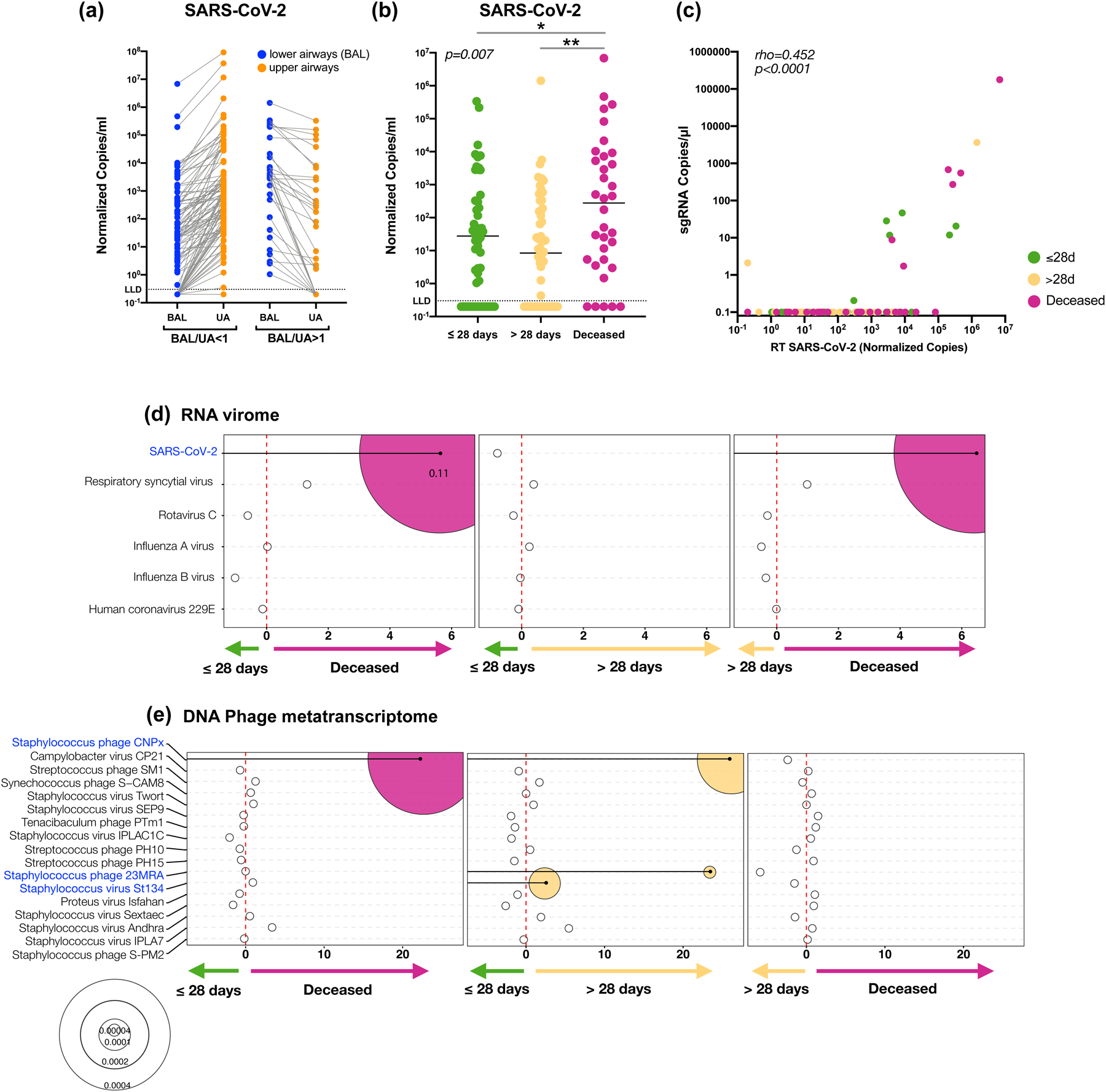
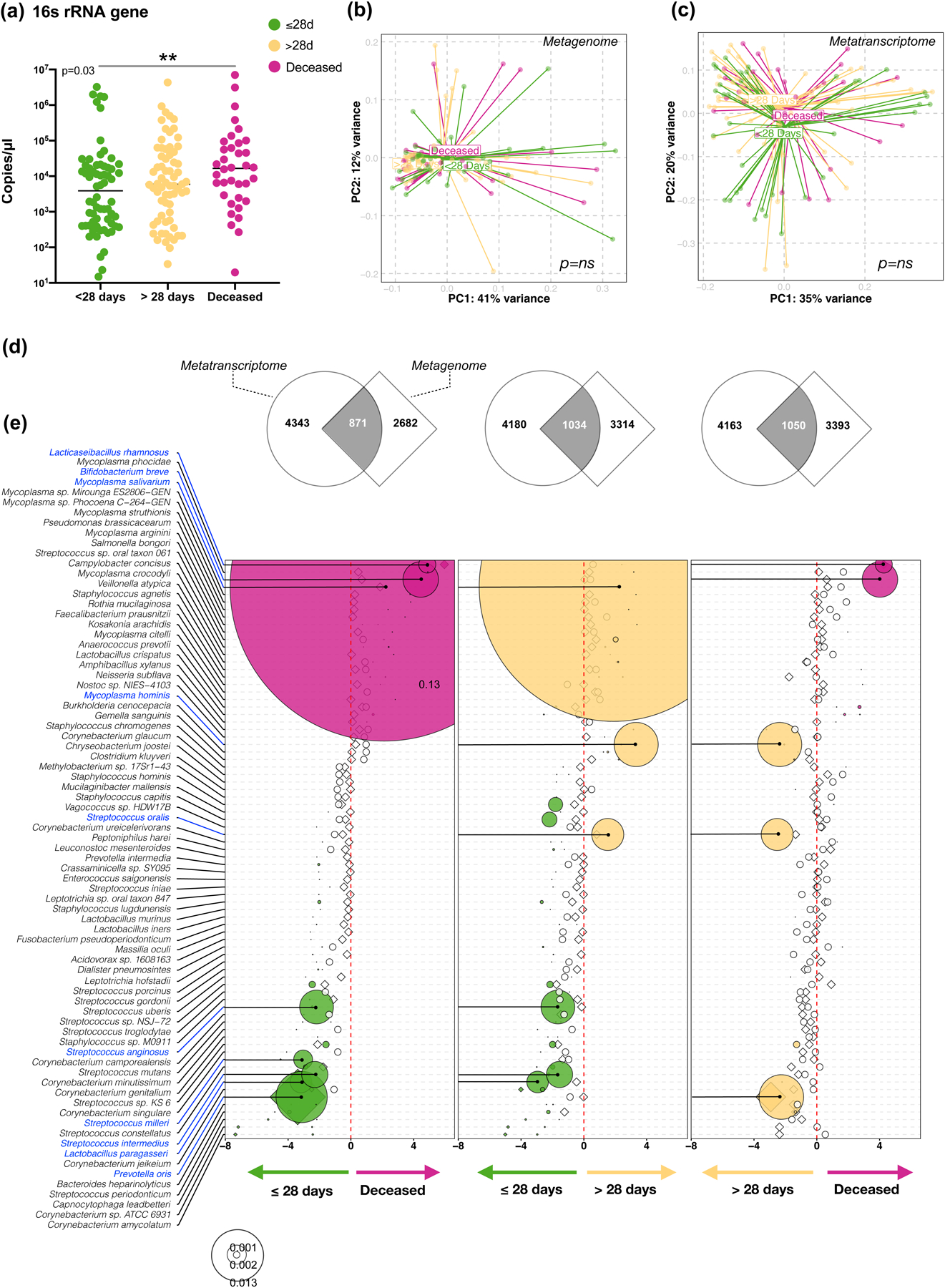
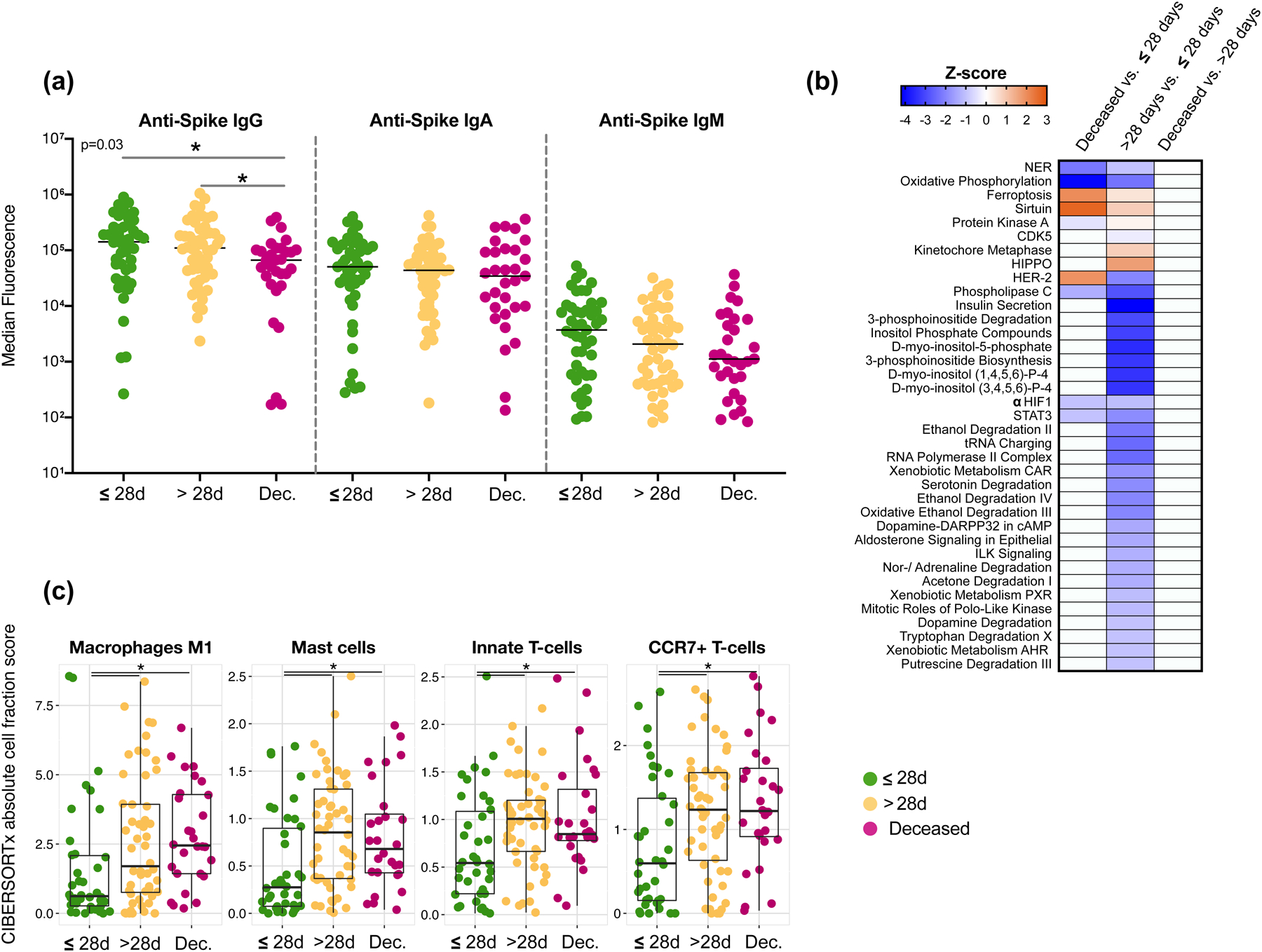
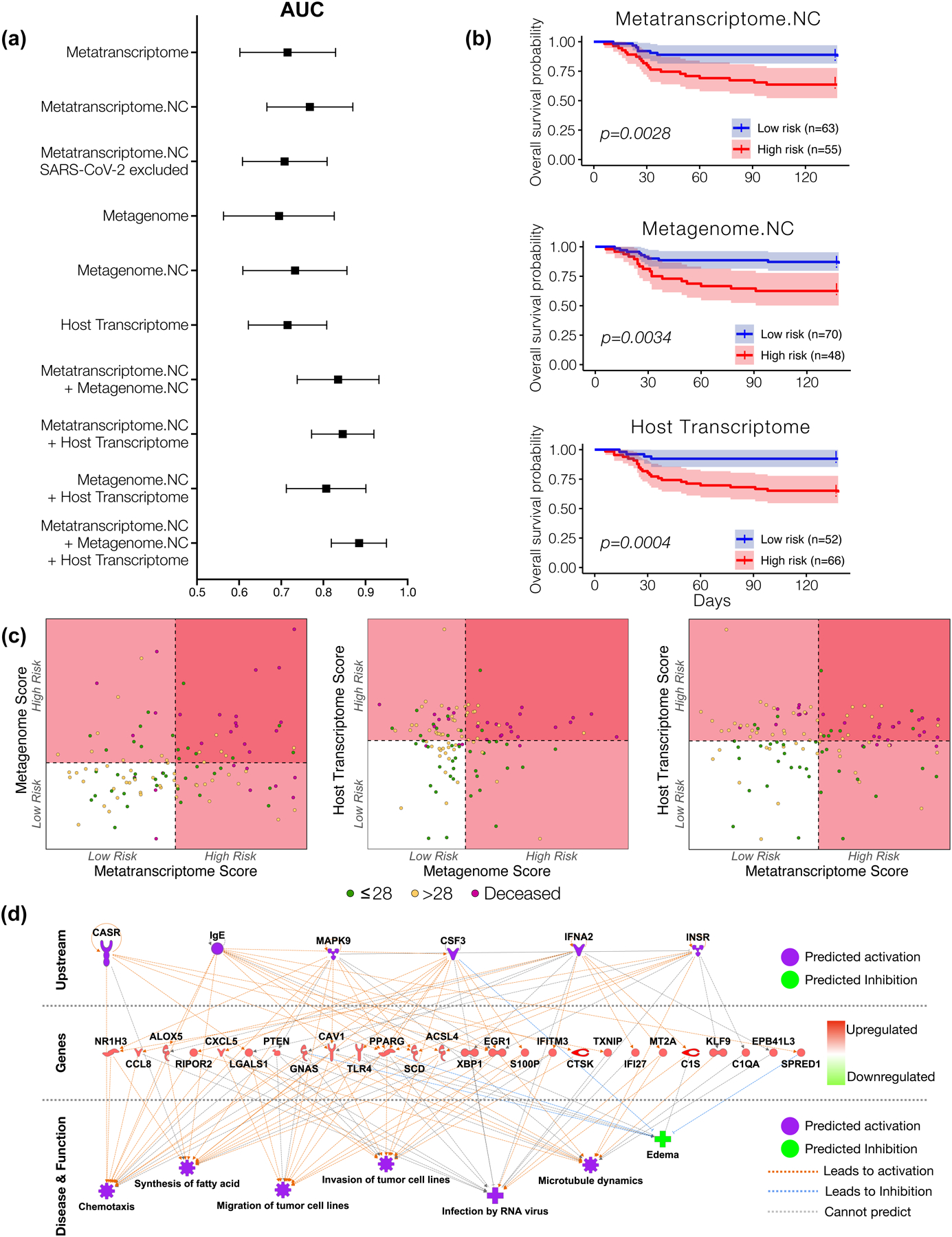
Respiratory failure is associated with increased mortality in COVID-19 patients. There are no validated lower airway biomarkers to predict clinical outcome. We investigated whether bacterial respiratory infections were associated with poor clinical outcome of COVID-19 in a prospective, observational cohort of 589 critically ill adults, all of whom required mechanical ventilation. For a subset of 142 patients who underwent bronchoscopy, we quantified SARS-CoV-2 viral load, analysed the lower respiratory tract microbiome using metagenomics and metatranscriptomics and profiled the host immune response. Acquisition of a hospital-acquired respiratory pathogen was not associated with fatal outcome. Poor clinical outcome was associated with lower airway enrichment with an oral commensal (Mycoplasma salivarium). Increased SARS-CoV-2 abundance, low anti-SARS-CoV-2 antibody response and a distinct host transcriptome profile of the lower airways were most predictive of mortality. Our data provide evidence that secondary respiratory infections do not drive mortality in COVID-19 and clinical management strategies should prioritize reducing viral replication and maximizing host responses to SARS-CoV-2.
© 2021. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing Interests Statement:
The authors declare no competing interests. The authors have no financial or non-financial interests to disclose.
Figures

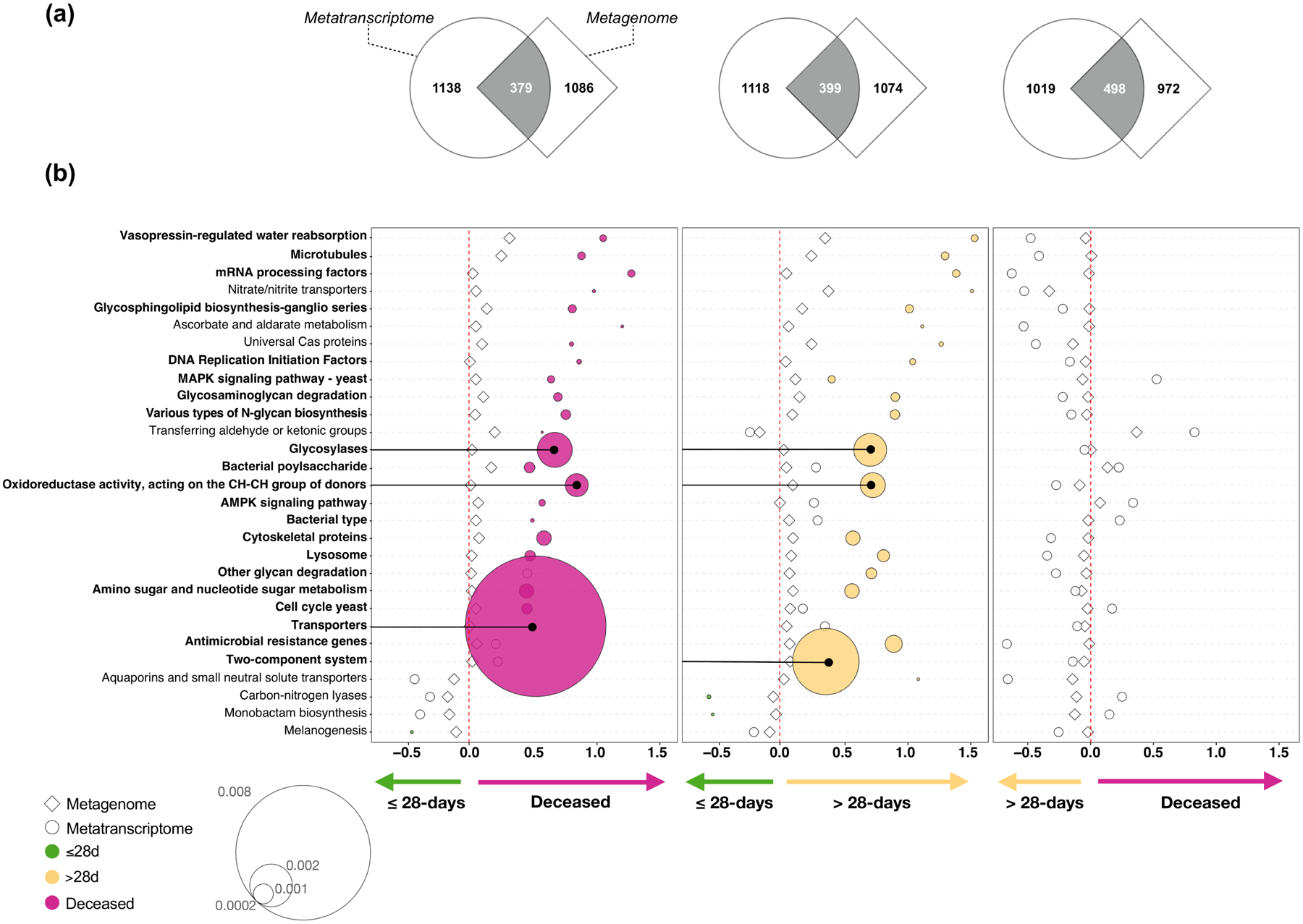
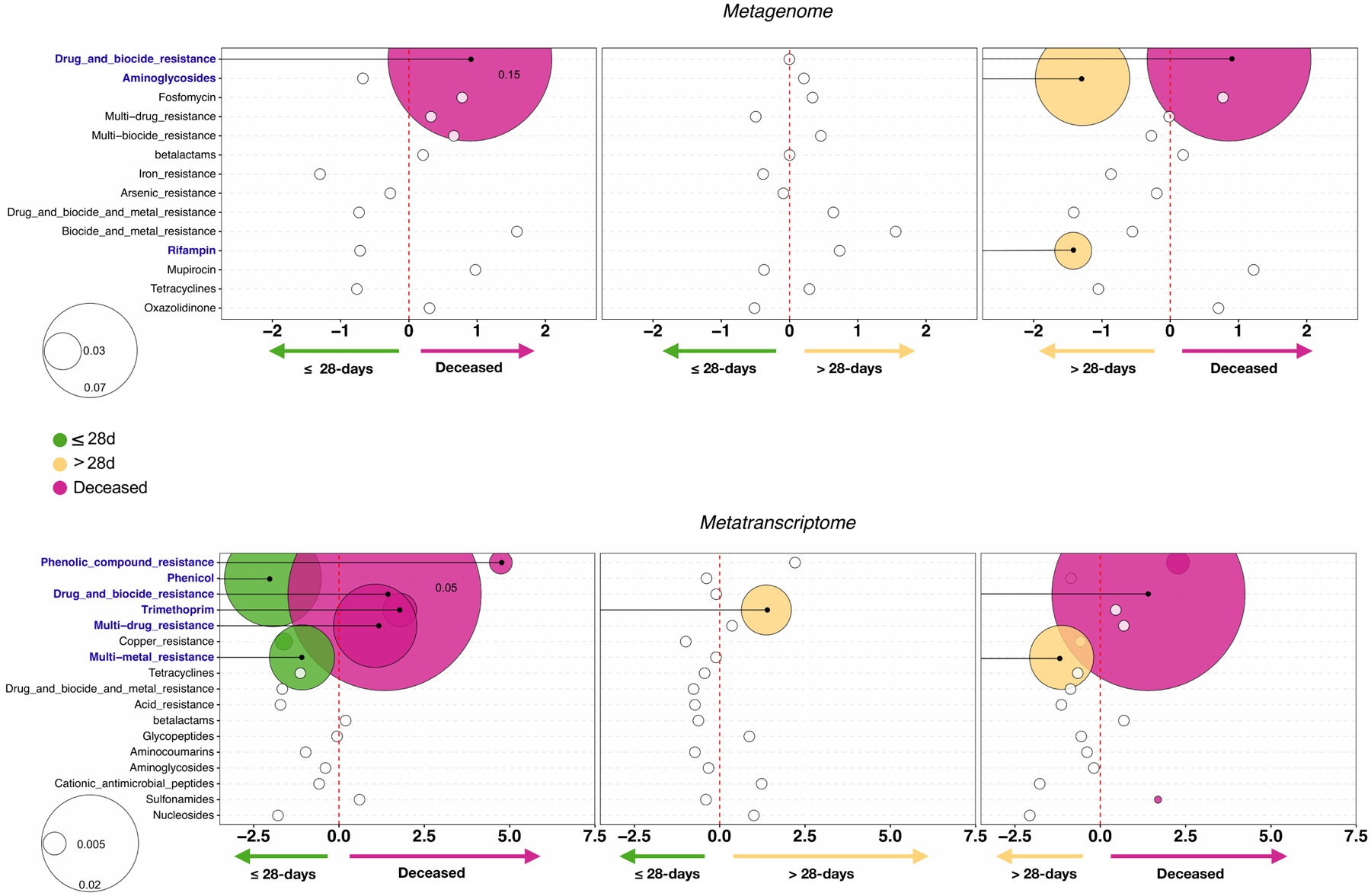
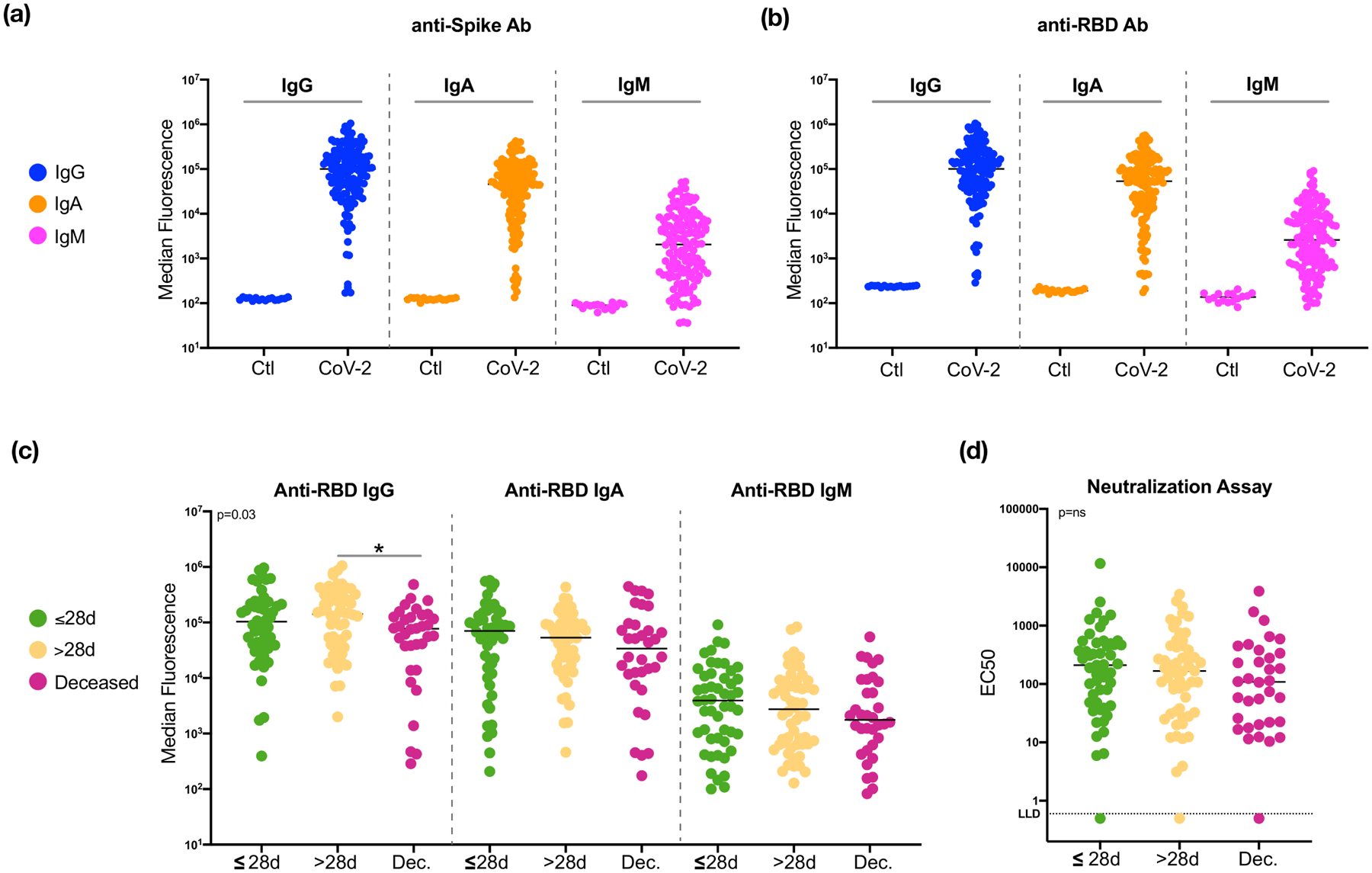
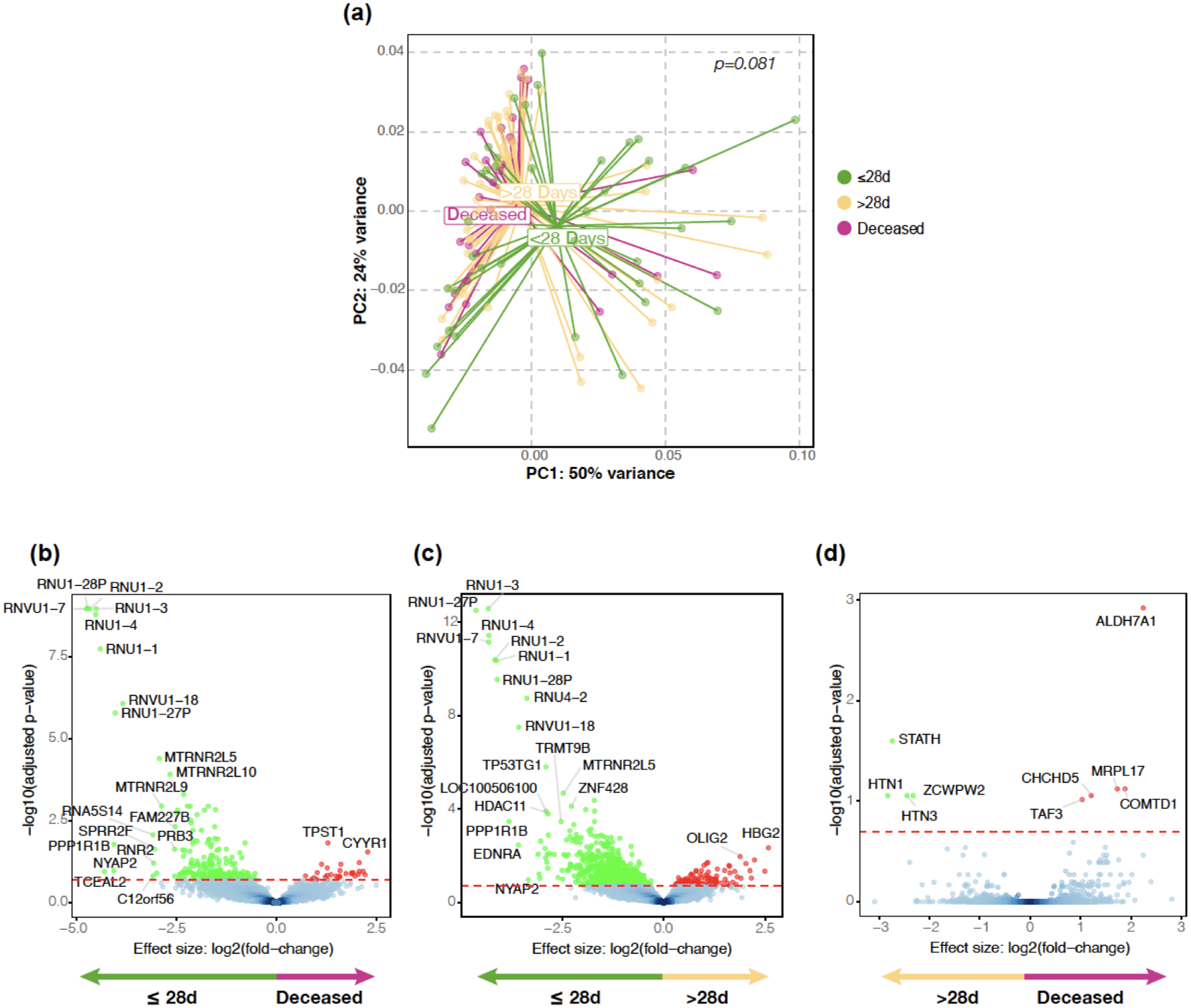
Update of
-
Microbial signatures in the lower airways of mechanically ventilated COVID19 patients associated with poor clinical outcome.medRxiv [Preprint]. 2021 Feb 26:2021.02.23.21252221. doi: 10.1101/2021.02.23.21252221. medRxiv. 2021. Update in: Nat Microbiol. 2021 Oct;6(10):1245-1258. doi: 10.1038/s41564-021-00961-5. PMID: 33655261 Free PMC article. Updated. Preprint.
-
Microbial signatures in the lower airways of mechanically ventilated COVID19 patients associated with poor clinical outcome.Res Sq [Preprint]. 2021 Mar 29:rs.3.rs-266050. doi: 10.21203/rs.3.rs-266050/v1. Res Sq. 2021. Update in: Nat Microbiol. 2021 Oct;6(10):1245-1258. doi: 10.1038/s41564-021-00961-5. PMID: 33791687 Free PMC article. Updated. Preprint.
References
-
- WHO coronavirus disease (COVID-19) dashboard. Geneva: World Health Organization, Available online: https://covid19.who.int/ (2020).
-
- Rabaan AA, et al.SARS-CoV-2, SARS-CoV, and MERS-COV: A comparative overview. Infez Med 28, 174–184 (2020). - PubMed
-
- Morens DM & Fauci AS The 1918 influenza pandemic: insights for the 21st century. J Infect Dis 195, 1018–1028 (2007). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
