

LZerD Protein-Protein Docking Webserver Enhanced With de novo Structure Prediction
- PMID: 34466411
- PMCID: PMC8403062
- DOI: 10.3389/fmolb.2021.724947
LZerD Protein-Protein Docking Webserver Enhanced With de novo Structure Prediction
Abstract
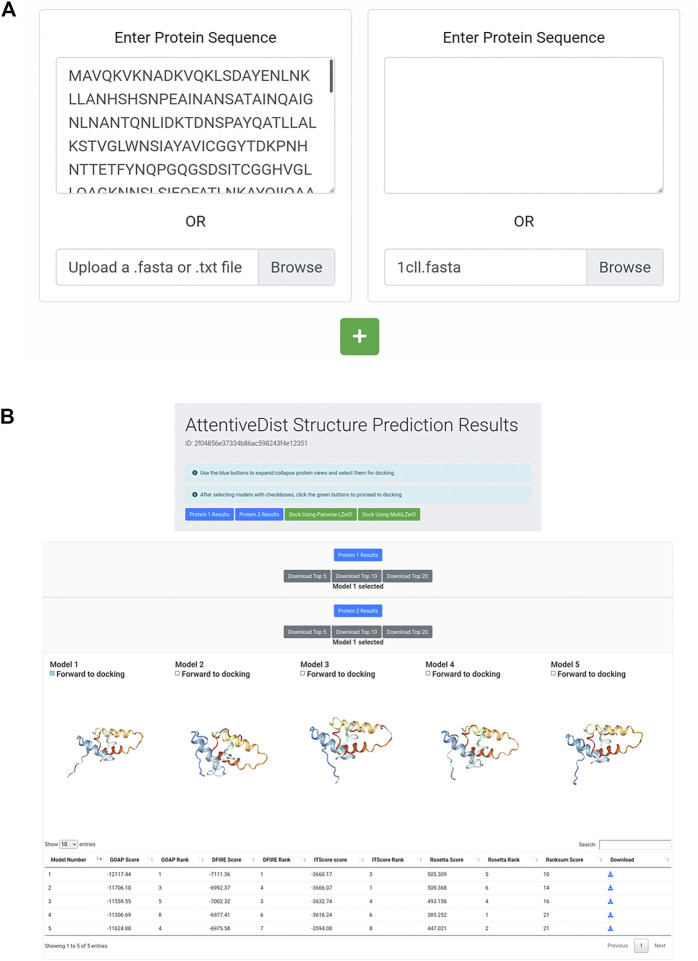
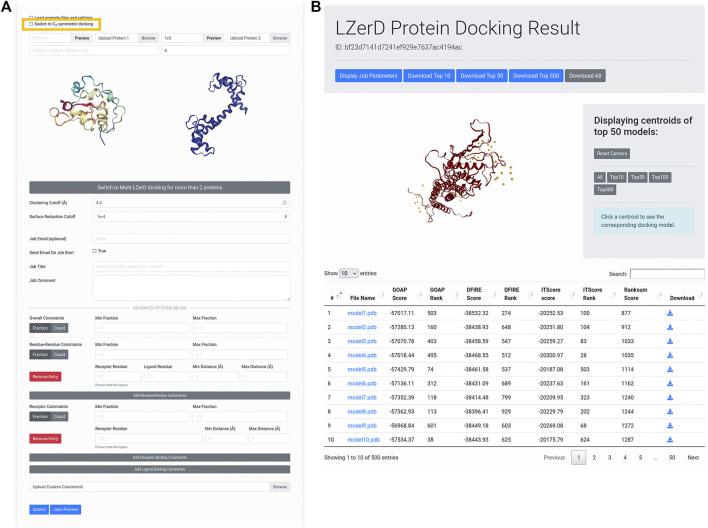
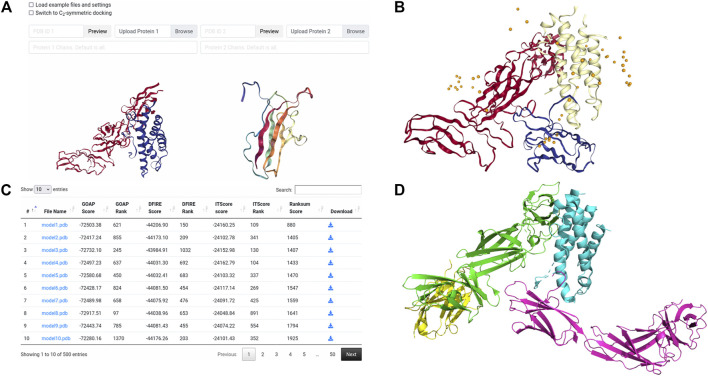
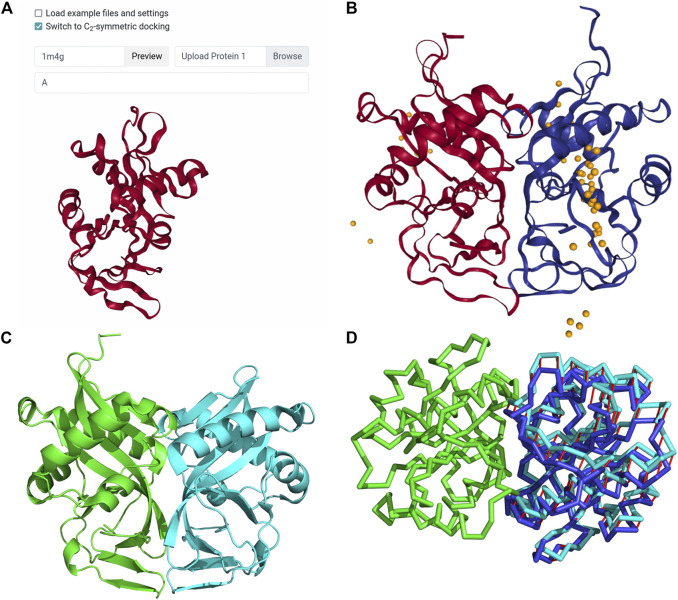
Protein-protein docking is a useful tool for modeling the structures of protein complexes that have yet to be experimentally determined. Understanding the structures of protein complexes is a key component for formulating hypotheses in biophysics regarding the functional mechanisms of complexes. Protein-protein docking is an established technique for cases where the structures of the subunits have been determined. While the number of known structures deposited in the Protein Data Bank is increasing, there are still many cases where the structures of individual proteins that users want to dock are not determined yet. Here, we have integrated the AttentiveDist method for protein structure prediction into our LZerD webserver for protein-protein docking, which enables users to simply submit protein sequences and obtain full-complex atomic models, without having to supply any structure themselves. We have further extended the LZerD docking interface with a symmetrical homodimer mode. The LZerD server is available at https://lzerd.kiharalab.org/.
Keywords: LZerD; protein bioinformatics; protein structure prediction; protein-protein docking; structure modeling; symmetrical docking; web server.
Copyright © 2021 Christoffer, Bharadwaj, Luu and Kihara.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources