

EpiScanpy: integrated single-cell epigenomic analysis
- PMID: 34471111
- PMCID: PMC8410937
- DOI: 10.1038/s41467-021-25131-3
EpiScanpy: integrated single-cell epigenomic analysis
Abstract
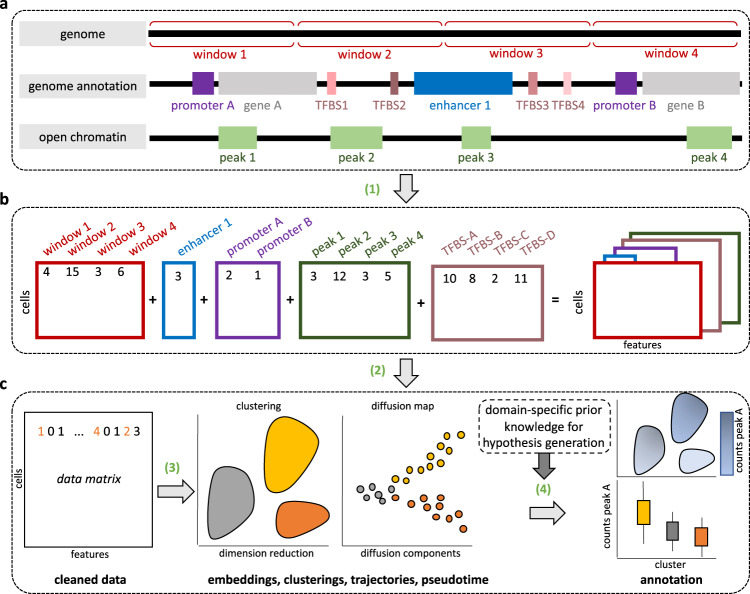
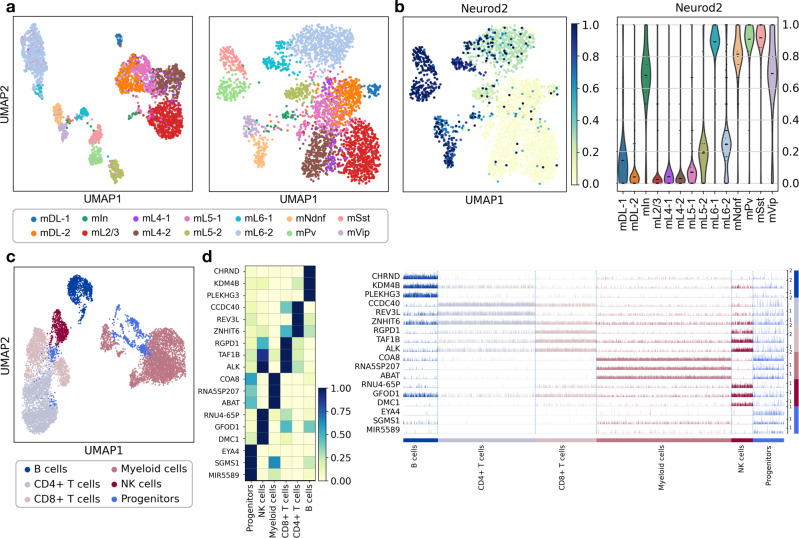
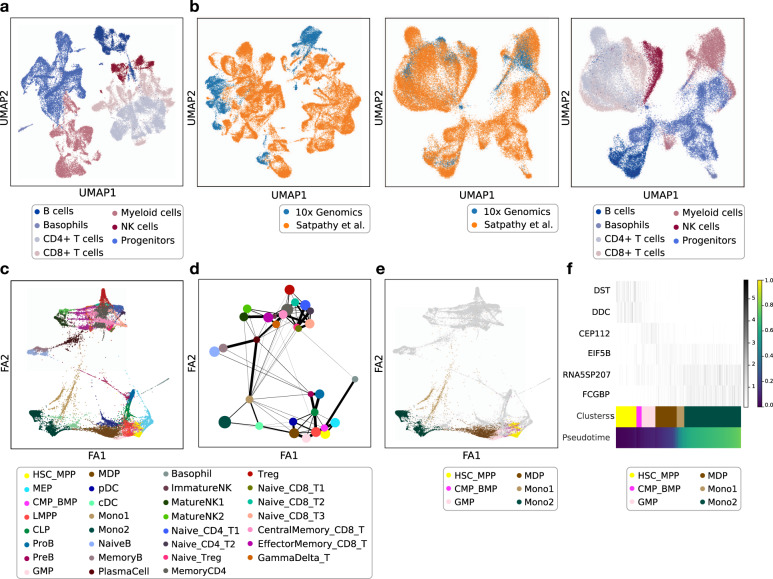
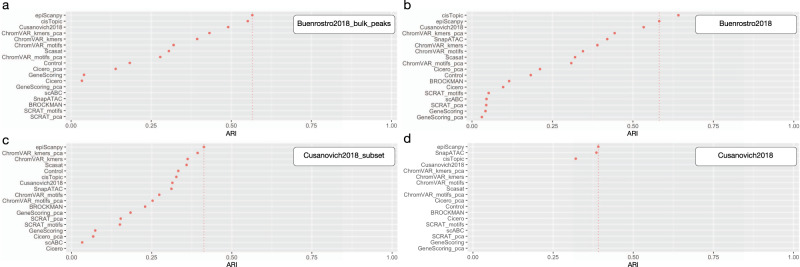
EpiScanpy is a toolkit for the analysis of single-cell epigenomic data, namely single-cell DNA methylation and single-cell ATAC-seq data. To address the modality specific challenges from epigenomics data, epiScanpy quantifies the epigenome using multiple feature space constructions and builds a nearest neighbour graph using epigenomic distance between cells. EpiScanpy makes the many existing scRNA-seq workflows from scanpy available to large-scale single-cell data from other -omics modalities, including methods for common clustering, dimension reduction, cell type identification and trajectory learning techniques, as well as an atlas integration tool for scATAC-seq datasets. The toolkit also features numerous useful downstream functions, such as differential methylation and differential openness calling, mapping epigenomic features of interest to their nearest gene, or constructing gene activity matrices using chromatin openness. We successfully benchmark epiScanpy against other scATAC-seq analysis tools and show its outperformance at discriminating cell types.
© 2021. The Author(s).
Conflict of interest statement
F.J.T. reports receiving consulting fees from Roche Diagnostics GmbH and Cellarity Inc., and ownership interest in Cellarity, Inc.. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
