

Highly diverse flavobacterial phages isolated from North Sea spring blooms
- PMID: 34475519
- PMCID: PMC8776804
- DOI: 10.1038/s41396-021-01097-4
Highly diverse flavobacterial phages isolated from North Sea spring blooms
Abstract
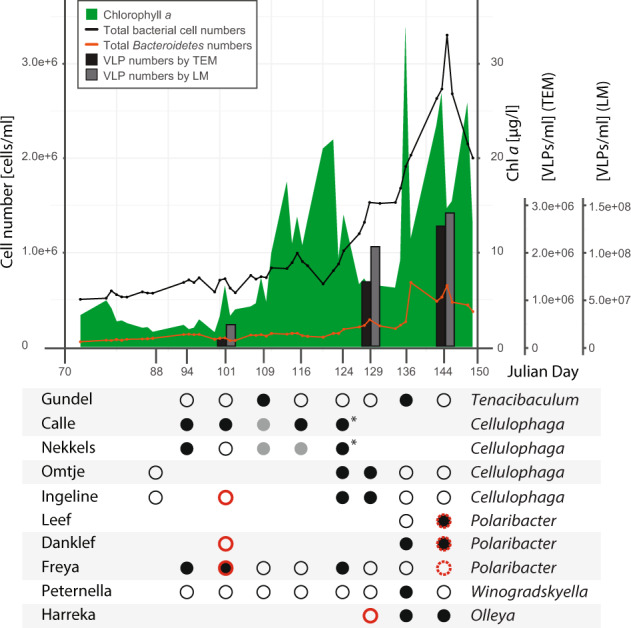
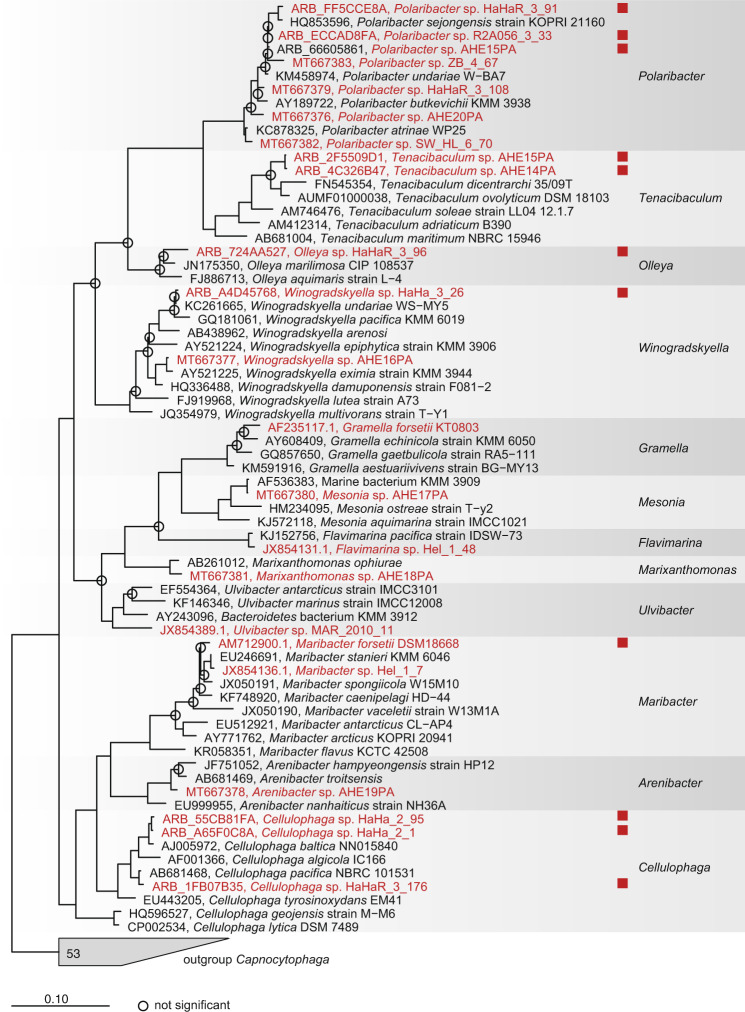
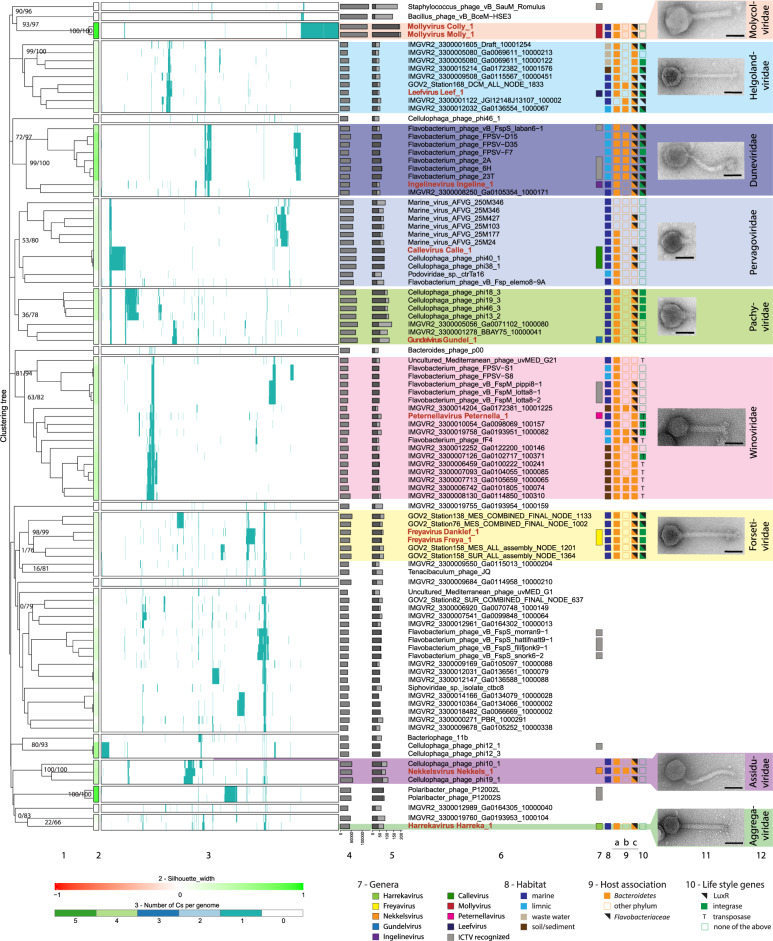
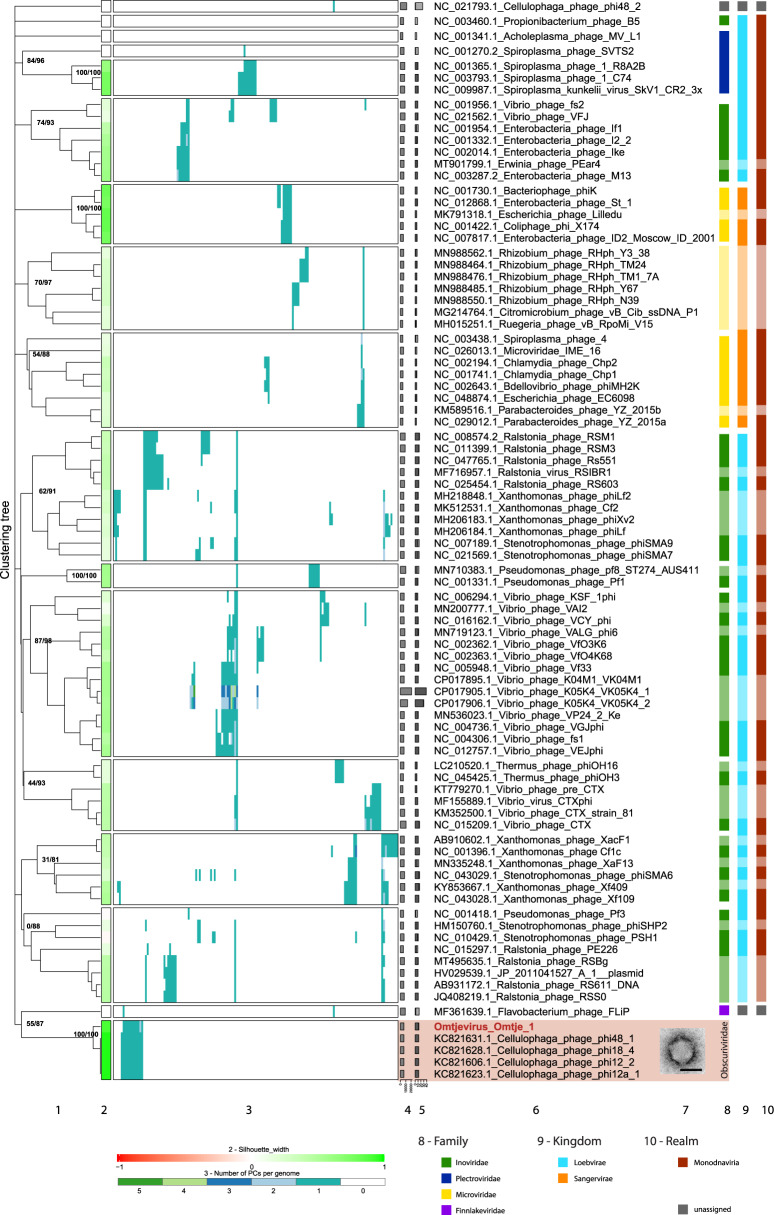
It is generally recognized that phages are a mortality factor for their bacterial hosts. This could be particularly true in spring phytoplankton blooms, which are known to be closely followed by a highly specialized bacterial community. We hypothesized that phages modulate these dense heterotrophic bacteria successions following phytoplankton blooms. In this study, we focused on Flavobacteriia, because they are main responders during these blooms and have an important role in the degradation of polysaccharides. A cultivation-based approach was used, obtaining 44 lytic flavobacterial phages (flavophages), representing twelve new species from two viral realms. Taxonomic analysis allowed us to delineate ten new phage genera and ten new families, from which nine and four, respectively, had no previously cultivated representatives. Genomic analysis predicted various life styles and genomic replication strategies. A likely eukaryote-associated host habitat was reflected in the gene content of some of the flavophages. Detection in cellular metagenomes and by direct-plating showed that part of these phages were actively replicating in the environment during the 2018 spring bloom. Furthermore, CRISPR/Cas spacers and re-isolation during two consecutive years suggested that, at least part of the new flavophages are stable components of the microbial community in the North Sea. Together, our results indicate that these diverse flavophages have the potential to modulate their respective host populations.
© 2021. The Author(s).
Conflict of interest statement
EMA is the current Chair of the Bacterial and Archaeal Viruses Subcommittee of the International Committee on Taxonomy of Viruses, and member of its Executive Committee. The other authors declare no competing interests.
Figures

References
-
- Proctor LM, Fuhrman JA. Viral mortality of marine bacteria and cyanobacteria. Nature. 1990;343:60–2.
-
- Steward G, Wikner J, Cochlan W, Smith D, Azam F. Estimation of virus production in the sea: II. field results. Mar Microb Food Webs. 1992;6:79–90.
-
- Suttle CA. The significance of viruses to mortality in aquatic microbial communities. Microb Ecol. 1994;28:237–43. - PubMed
-
- Thingstad TF. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol Oceanogr. 2000;45:1320–8.
-
- Wilhelm SW, Suttle CA. Viruses and nutrient cycles in the sea: viruses play critical roles in the structure and function of aquatic food webs. BioScience. 1999;49:781–8.
