

Fast two-stage phasing of large-scale sequence data
- PMID: 34478634
- PMCID: PMC8551421
- DOI: 10.1016/j.ajhg.2021.08.005
Fast two-stage phasing of large-scale sequence data
Abstract
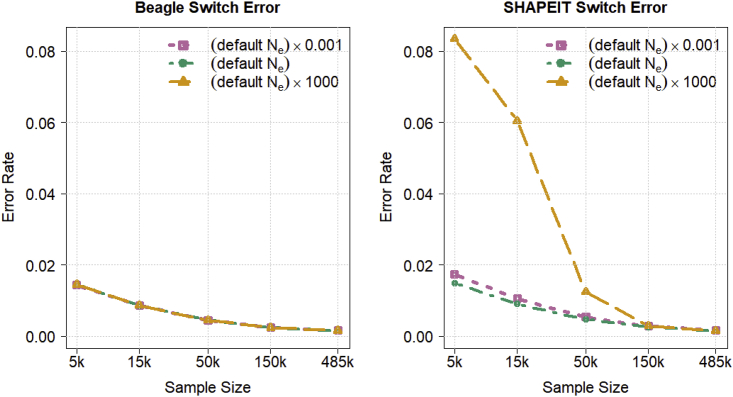
Haplotype phasing is the estimation of haplotypes from genotype data. We present a fast, accurate, and memory-efficient haplotype phasing method that scales to large-scale SNP array and sequence data. The method uses marker windowing and composite reference haplotypes to reduce memory usage and computation time. It incorporates a progressive phasing algorithm that identifies confidently phased heterozygotes in each iteration and fixes the phase of these heterozygotes in subsequent iterations. For data with many low-frequency variants, such as whole-genome sequence data, the method employs a two-stage phasing algorithm that phases high-frequency markers via progressive phasing in the first stage and phases low-frequency markers via genotype imputation in the second stage. This haplotype phasing method is implemented in the open-source Beagle 5.2 software package. We compare Beagle 5.2 and SHAPEIT 4.2.1 by using expanding subsets of 485,301 UK Biobank samples and 38,387 TOPMed samples. Both methods have very similar accuracy and computation time for UK Biobank SNP array data. However, for TOPMed sequence data, Beagle is more than 20 times faster than SHAPEIT, achieves similar accuracy, and scales to larger sample sizes.
Keywords: TOPMed; UK Biobank; genotype phasing; haplotype phasing; phasing.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
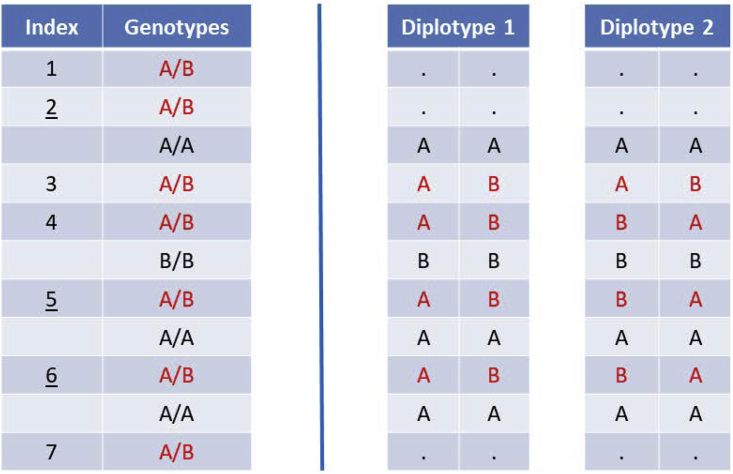
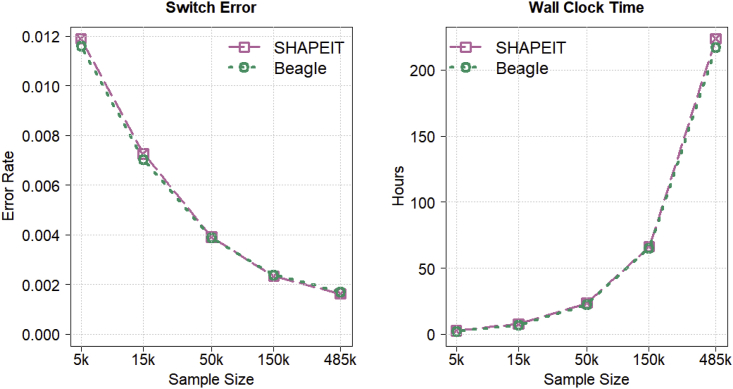
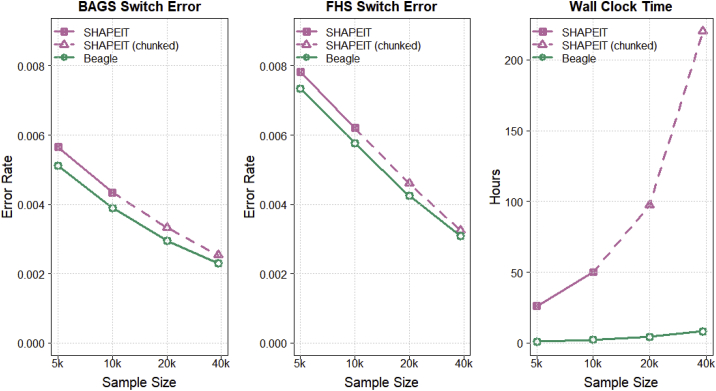
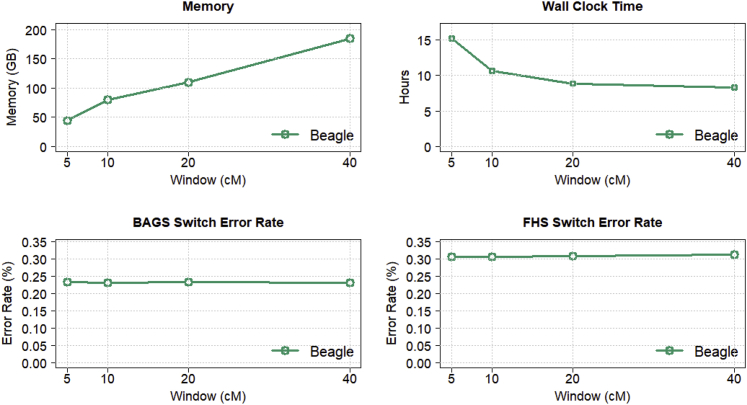
References
-
- Das S., Abecasis G.R., Browning B.L. Genotype Imputation from Large Reference Panels. Annu. Rev. Genomics Hum. Genet. 2018;19:73–96. - PubMed
-
- Larsen L.A., Fosdal I., Andersen P.S., Kanters J.K., Vuust J., Wettrell G., Christiansen M. Recessive Romano-Ward syndrome associated with compound heterozygosity for two mutations in the KVLQT1 gene. Eur. J. Hum. Genet. 1999;7:724–728. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 HL120393/HL/NHLBI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- HHSN268201500001C/HL/NHLBI NIH HHS/United States
- U01 HL120393/HL/NHLBI NIH HHS/United States
- HHSN268201500001I/HL/NHLBI NIH HHS/United States
- R01 HL104608/HL/NHLBI NIH HHS/United States
- 75N92019D00031/HL/NHLBI NIH HHS/United States
- MC_PC_17228/MRC_/Medical Research Council/United Kingdom
- R01 HL092577/HL/NHLBI NIH HHS/United States
- MC_QA137853/MRC_/Medical Research Council/United Kingdom
- HHSN268201800001C/HL/NHLBI NIH HHS/United States
- R01 HL087699/HL/NHLBI NIH HHS/United States
- N01 HC025195/HL/NHLBI NIH HHS/United States
- R01 HL117626/HL/NHLBI NIH HHS/United States
- R01 HG008359/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
