

Systematic evaluation of chromosome conformation capture assays
- PMID: 34480151
- PMCID: PMC8446342
- DOI: 10.1038/s41592-021-01248-7
Systematic evaluation of chromosome conformation capture assays
Abstract
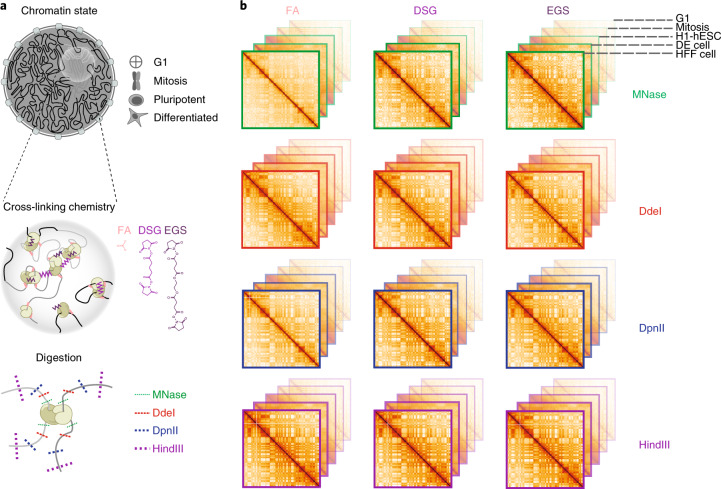
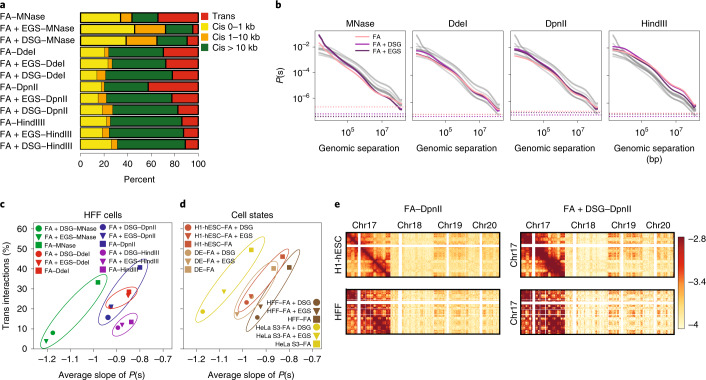
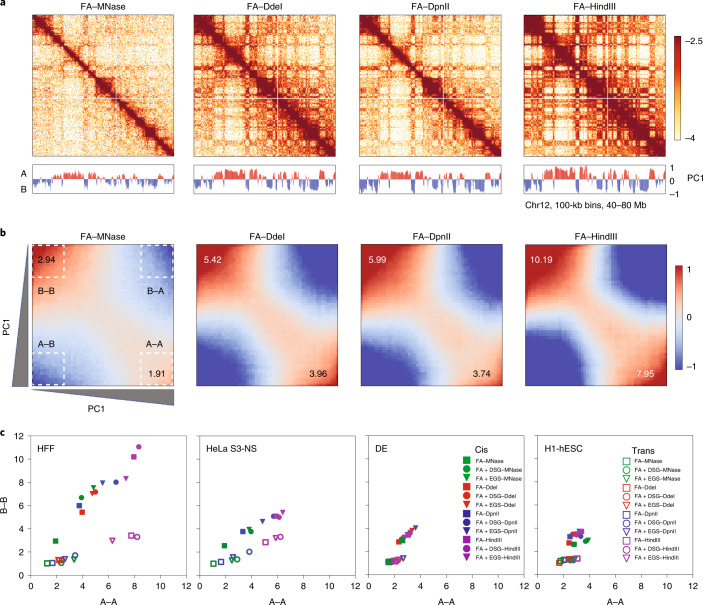
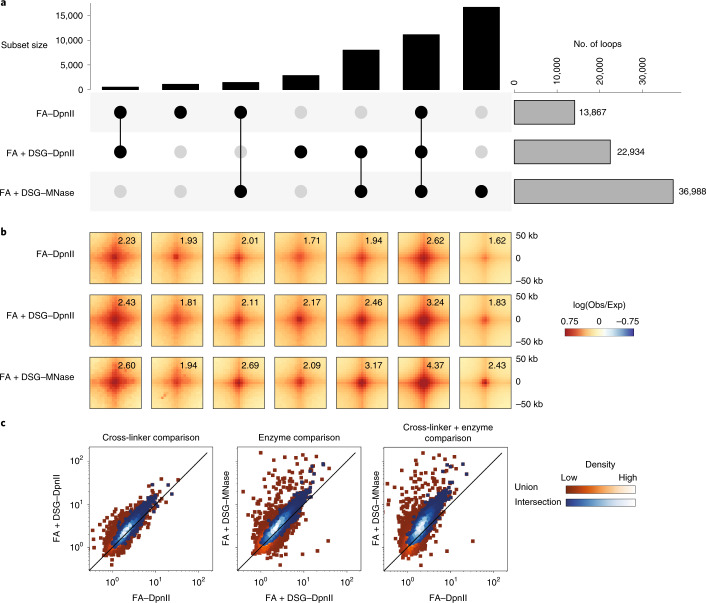
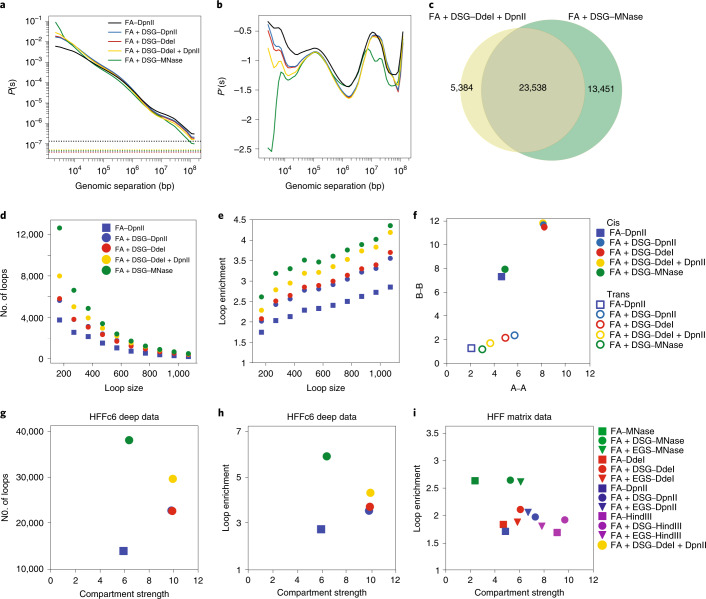
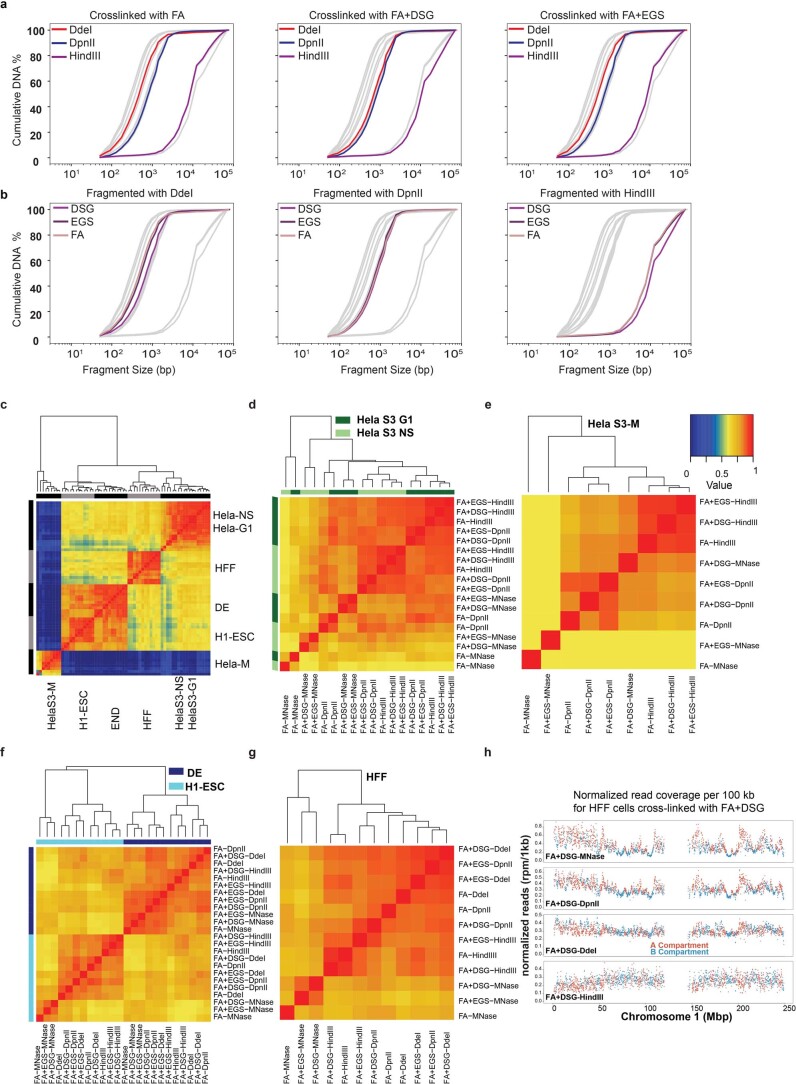
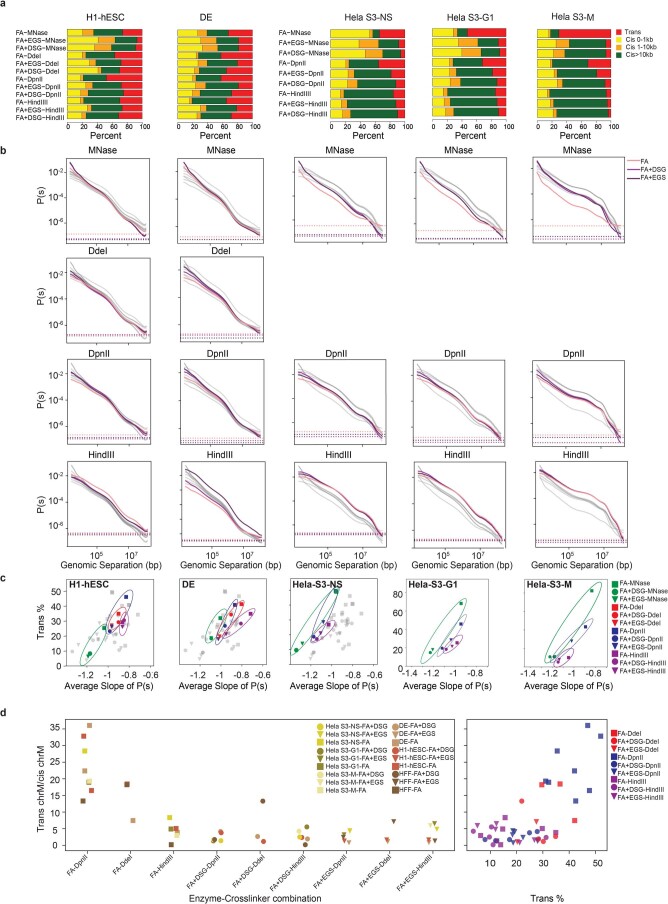
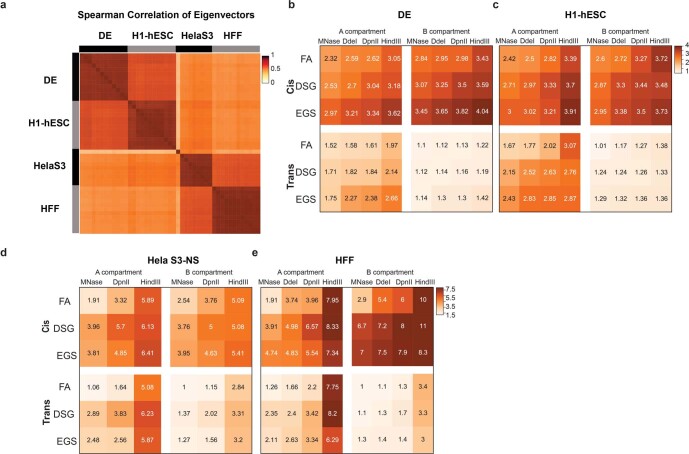
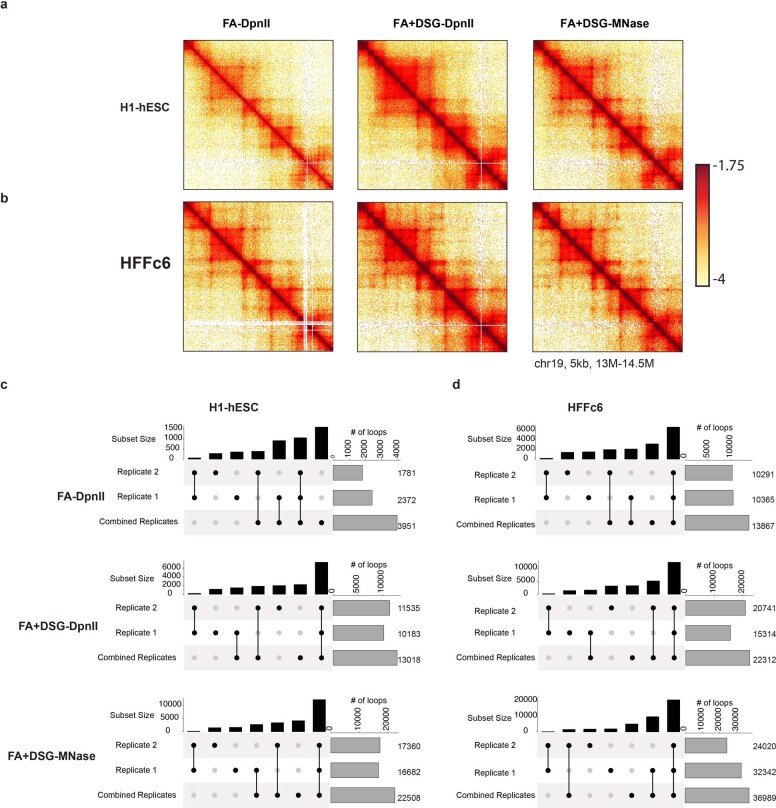
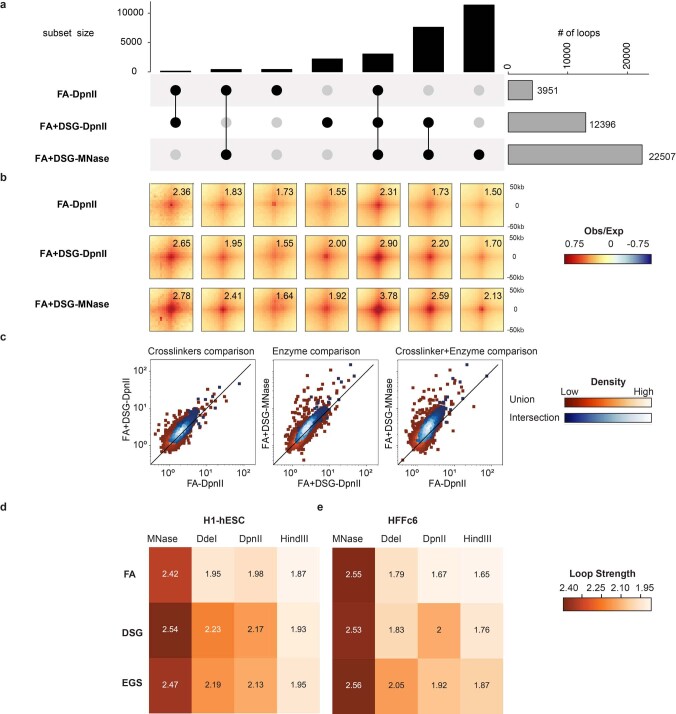
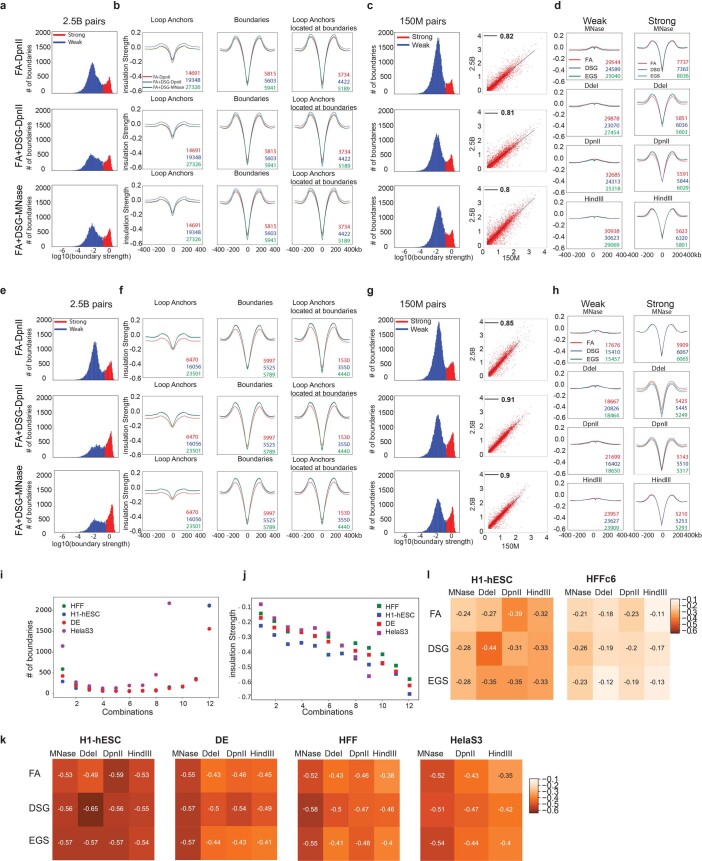
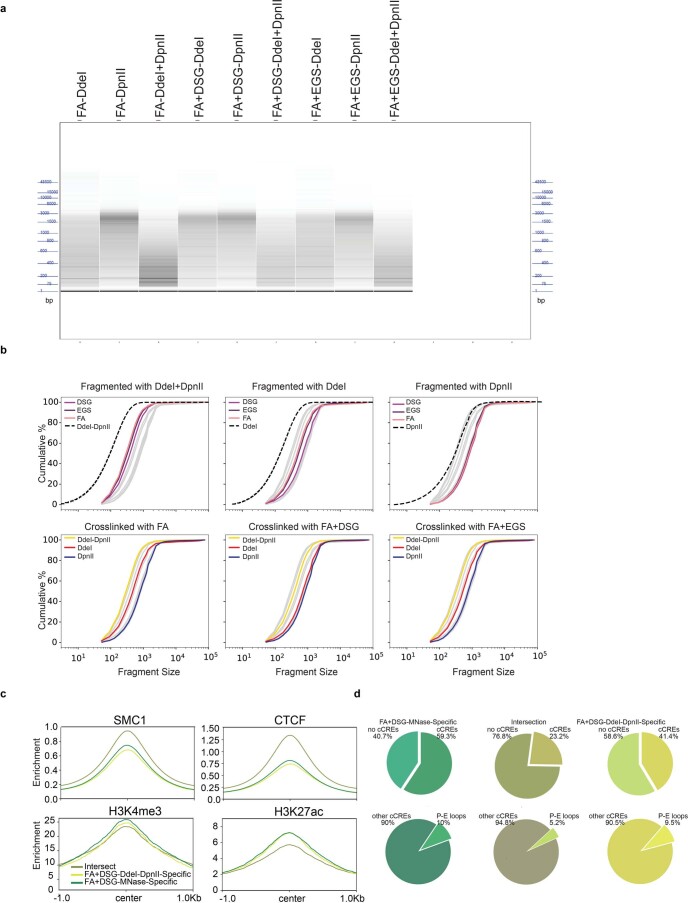
Chromosome conformation capture (3C) assays are used to map chromatin interactions genome-wide. Chromatin interaction maps provide insights into the spatial organization of chromosomes and the mechanisms by which they fold. Hi-C and Micro-C are widely used 3C protocols that differ in key experimental parameters including cross-linking chemistry and chromatin fragmentation strategy. To understand how the choice of experimental protocol determines the ability to detect and quantify aspects of chromosome folding we have performed a systematic evaluation of 3C experimental parameters. We identified optimal protocol variants for either loop or compartment detection, optimizing fragment size and cross-linking chemistry. We used this knowledge to develop a greatly improved Hi-C protocol (Hi-C 3.0) that can detect both loops and compartments relatively effectively. In addition to providing benchmarked protocols, this work produced ultra-deep chromatin interaction maps using Micro-C, conventional Hi-C and Hi-C 3.0 for key cell lines used by the 4D Nucleome project.
© 2021. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
