

Identification and prioritization of myeloid malignancy germline variants in a large cohort of adult patients with AML
- PMID: 34482403
- PMCID: PMC9211447
- DOI: 10.1182/blood.2021011354
Identification and prioritization of myeloid malignancy germline variants in a large cohort of adult patients with AML
Abstract
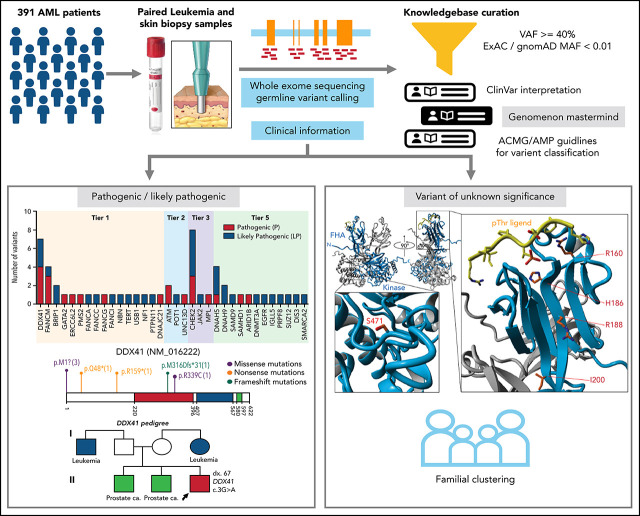
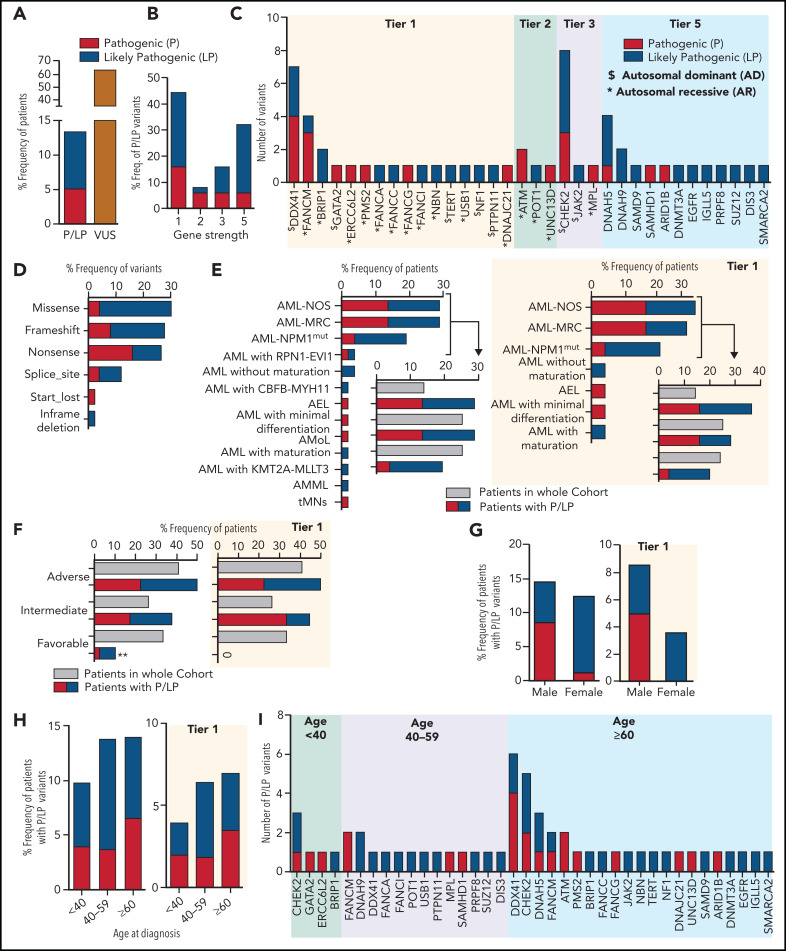
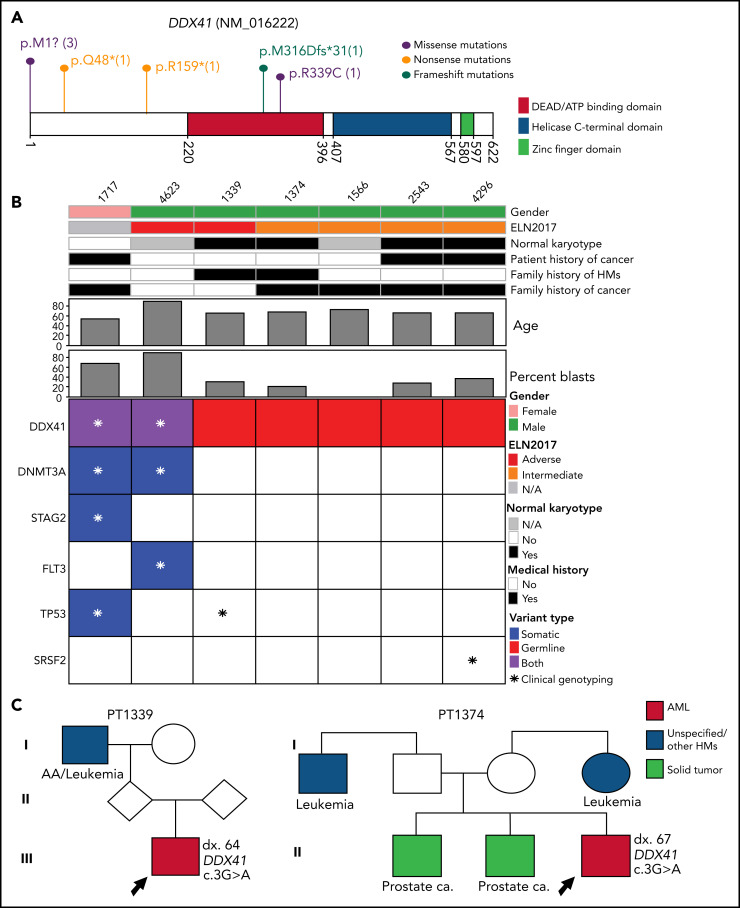
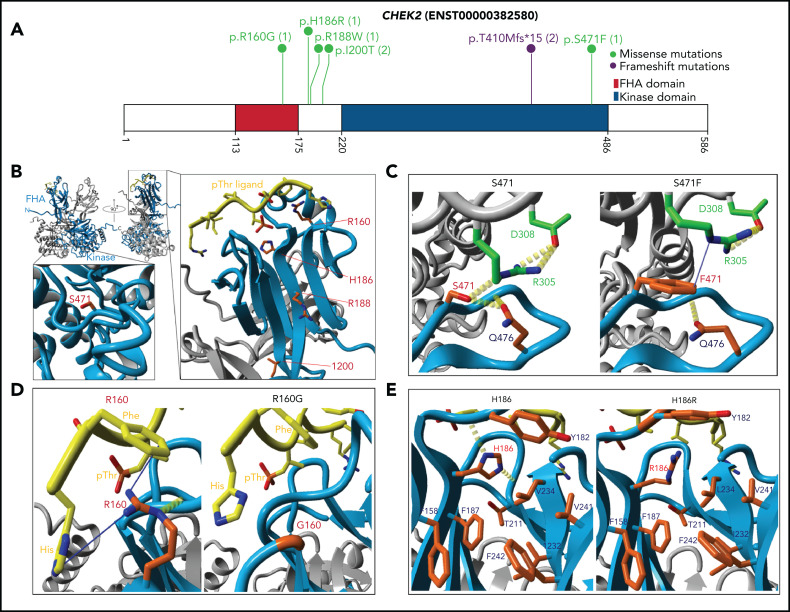
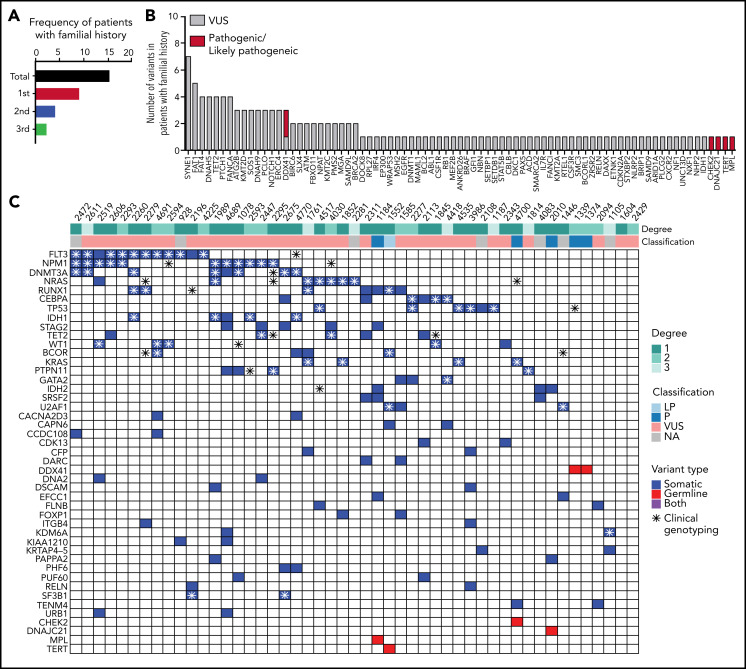
Inherited predisposition to myeloid malignancies is more common than previously appreciated. We analyzed the whole-exome sequencing data of paired leukemia and skin biopsy samples from 391 adult patients from the Beat AML 1.0 consortium. Using the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines for variant interpretation, we curated 1547 unique variants from 228 genes. The pathogenic/likely pathogenic (P/LP) germline variants were identified in 53 acute myeloid leukemia (AML) patients (13.6%) in 34 genes, including 6.39% (25/391) of patients harboring P/LP variants in genes considered clinically actionable (tier 1). 41.5% of the 53 patients with P/LP variants were in genes associated with the DNA damage response. The most frequently mutated genes were CHEK2 (8 patients) and DDX41 (7 patients). Pathogenic germline variants were also found in new candidate genes (DNAH5, DNAH9, DNMT3A, and SUZ12). No strong correlation was found between the germline mutational rate and age of AML onset. Among 49 patients who have a reported history of at least one family member affected with hematological malignancies, 6 patients harbored known P/LP germline variants and the remaining patients had at least one variant of uncertain significance, suggesting a need for further functional validation studies. Using CHEK2 as an example, we show that three-dimensional protein modeling can be one of the effective methodologies to prioritize variants of unknown significance for functional studies. Further, we evaluated an in silico approach that applies ACMG curation in an automated manner using the tool for assessment and (TAPES) prioritization in exome studies, which can minimize manual curation time for variants. Overall, our findings suggest a need to comprehensively understand the predisposition potential of many germline variants in order to enable closer monitoring for disease management and treatment interventions for affected patients and families.
© 2022 by The American Society of Hematology.
Figures

Comment in
-
And the germline beat (AML) goes on.Blood. 2022 Feb 24;139(8):1126-1128. doi: 10.1182/blood.2021013771. Blood. 2022. PMID: 35201331 No abstract available.
References
-
- Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136-1152. - PubMed
-
- Song WJ, Sullivan MG, Legare RD, et al. . Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166-175. - PubMed
-
- Wartiovaara-Kautto U, Hirvonen EAM, Pitkänen E, et al. . Germline alterations in a consecutive series of acute myeloid leukemia. Leukemia. 2018;32(10):2282-2285. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
