

Host-specific asymmetric accumulation of mutation types reveals that the origin of SARS-CoV-2 is consistent with a natural process
- PMID: 34485968
- PMCID: PMC8405235
- DOI: 10.1016/j.xinn.2021.100159
Host-specific asymmetric accumulation of mutation types reveals that the origin of SARS-CoV-2 is consistent with a natural process
Abstract
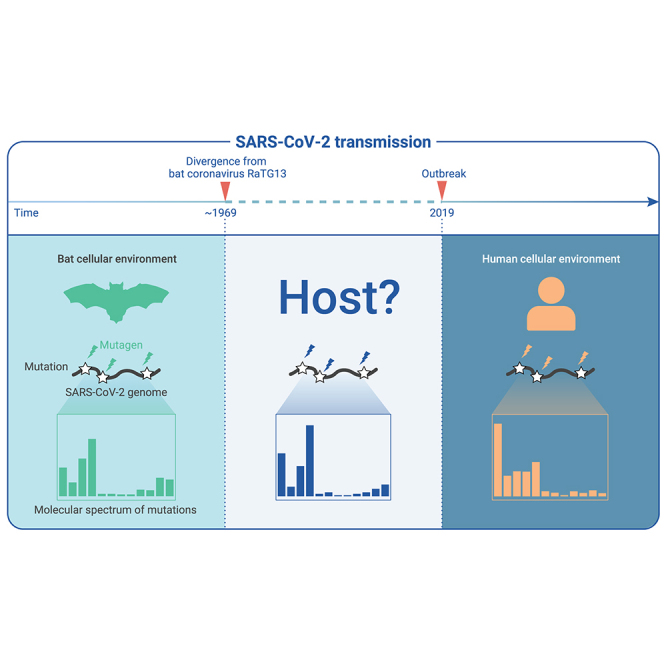
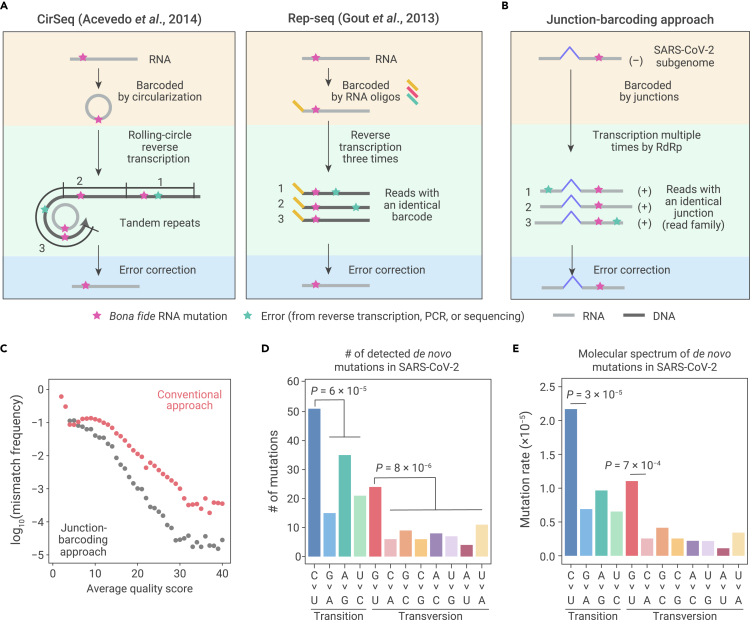
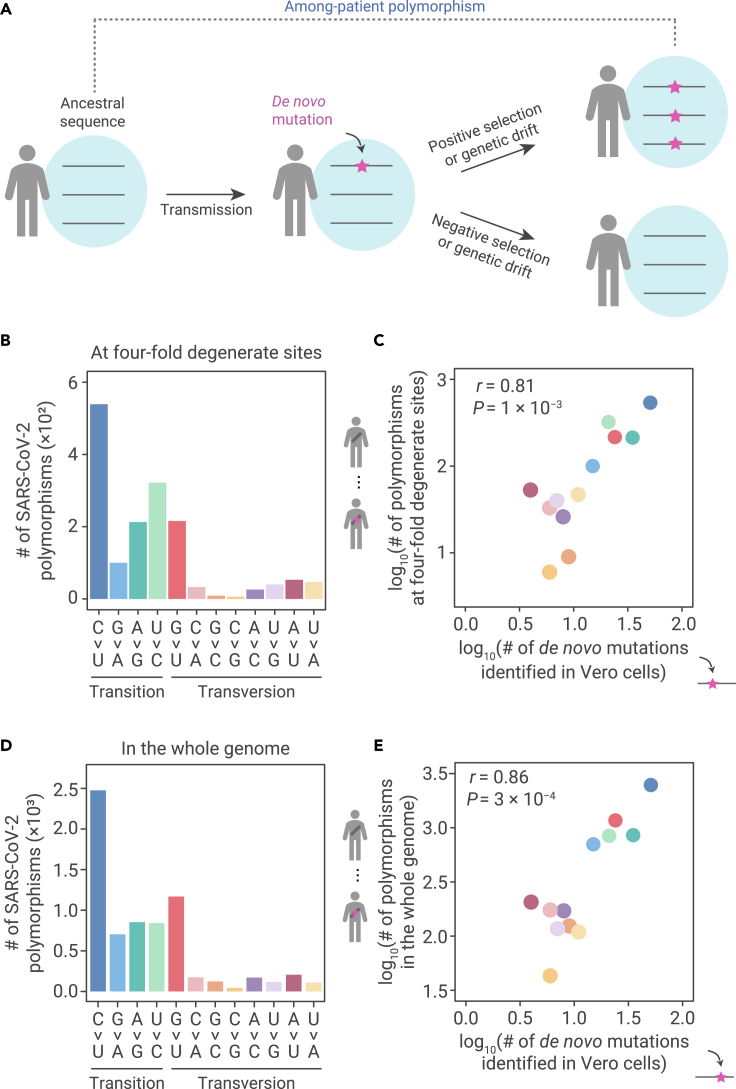
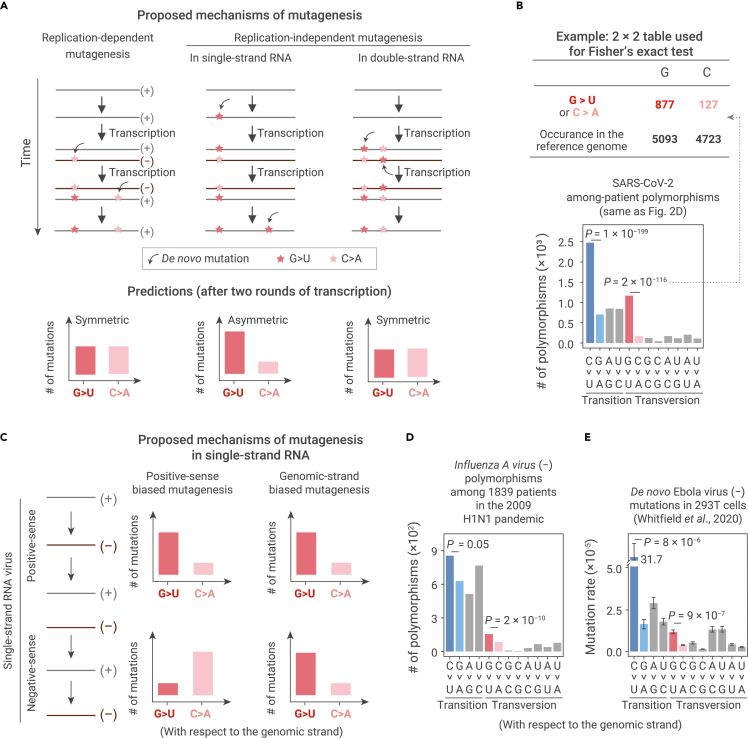
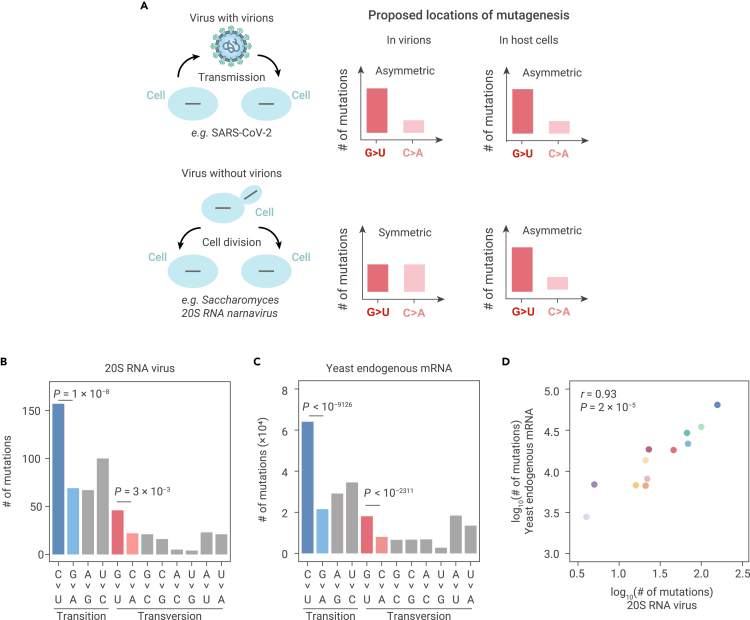
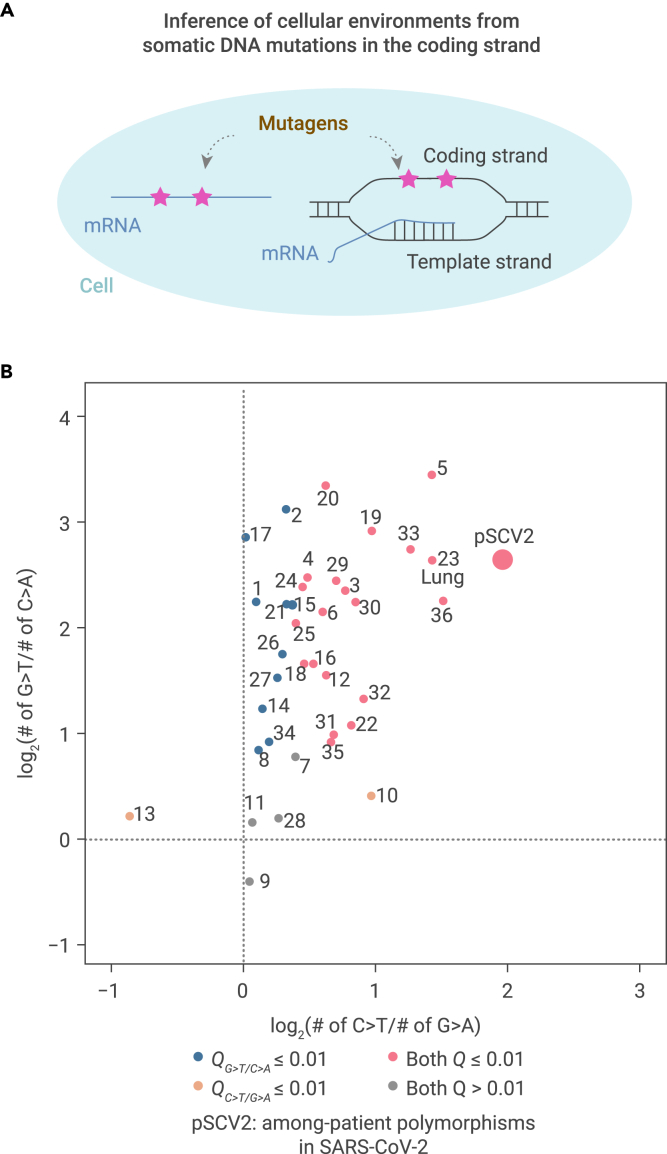
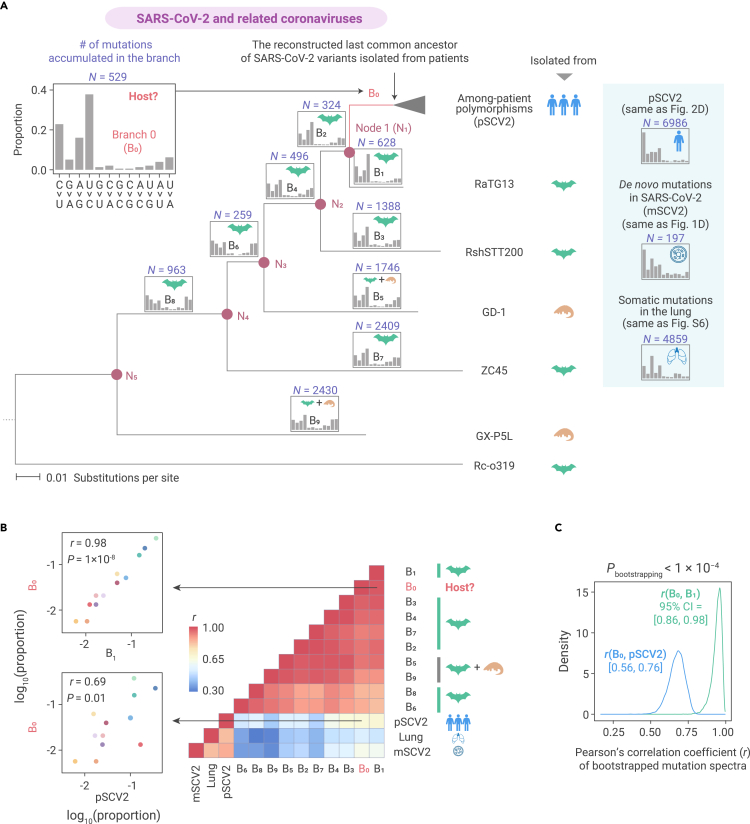
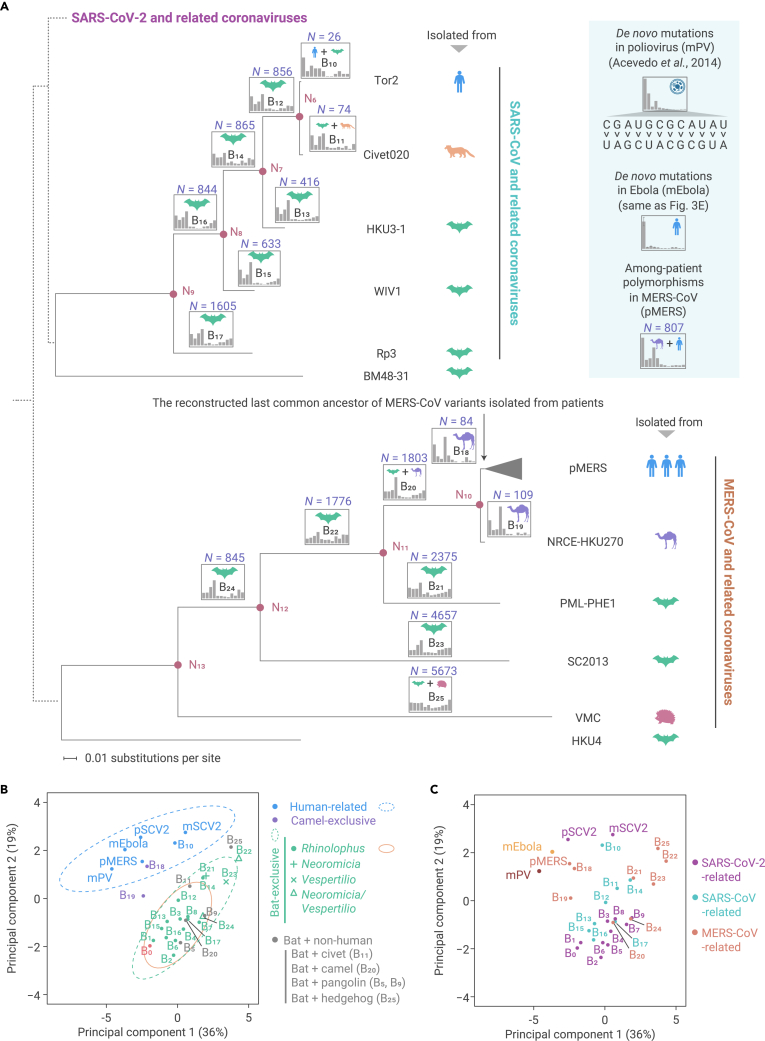
The capacity of RNA viruses to adapt to new hosts and rapidly escape the host immune system is largely attributable to de novo genetic diversity that emerges through mutations in RNA. Although the molecular spectrum of de novo mutations-the relative rates at which various base substitutions occur-are widely recognized as informative toward understanding the evolution of a viral genome, little attention has been paid to the possibility of using molecular spectra to infer the host origins of a virus. Here, we characterize the molecular spectrum of de novo mutations for SARS-CoV-2 from transcriptomic data obtained from virus-infected cell lines, enabled by the use of sporadic junctions formed during discontinuous transcription as molecular barcodes. We find that de novo mutations are generated in a replication-independent manner, typically on the genomic strand, and highly dependent on mutagenic mechanisms specific to the host cellular environment. De novo mutations will then strongly influence the types of base substitutions accumulated during SARS-CoV-2 evolution, in an asymmetric manner favoring specific mutation types. Consequently, similarities between the mutation spectra of SARS-CoV-2 and the bat coronavirus RaTG13, which have accumulated since their divergence strongly suggest that SARS-CoV-2 evolved in a host cellular environment highly similar to that of bats before its zoonotic transfer into humans. Collectively, our findings provide data-driven support for the natural origin of SARS-CoV-2.
Keywords: SARS-CoV-2; de novo mutations; evolutionary origin; mRNA mutation; molecular spectrum; mutational signature.
© 2021 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Rasmussen A.L. On the origins of SARS-CoV-2. Nat. Med. 2021;27:9. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
