

BleTIES: annotation of natural genome editing in ciliates using long read sequencing
- PMID: 34487139
- PMCID: PMC11301610
- DOI: 10.1093/bioinformatics/btab613
BleTIES: annotation of natural genome editing in ciliates using long read sequencing
Abstract
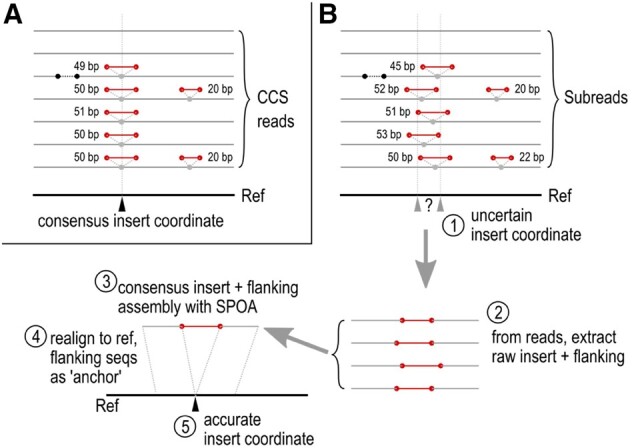
Summary: Ciliates are single-celled eukaryotes that eliminate specific, interspersed DNA sequences (internally eliminated sequences, IESs) from their genomes during development. These are challenging to annotate and assemble because IES-containing sequences are typically much less abundant in the cell than those without, and IES sequences themselves often contain repetitive and low-complexity sequences. Long-read sequencing technologies from Pacific Biosciences and Oxford Nanopore have the potential to reconstruct longer IESs than has been possible with short reads but require a different assembly strategy. Here we present BleTIES, a software toolkit for detecting, assembling, and analyzing IESs using mapped long reads.
Availability and implementation: BleTIES is implemented in Python 3. Source code is available at https://github.com/Swart-lab/bleties (MIT license) and also distributed via Bioconda.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2021. Published by Oxford University Press.
Figures
References
-
- Denby Wilkes C. et al. (2016) ParTIES: a toolbox for Paramecium interspersed DNA elimination studies. Bioinformatics, 32, 599–601. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
