

Gramtools enables multiscale variation analysis with genome graphs
- PMID: 34488837
- PMCID: PMC8420074
- DOI: 10.1186/s13059-021-02474-0
Gramtools enables multiscale variation analysis with genome graphs
Abstract
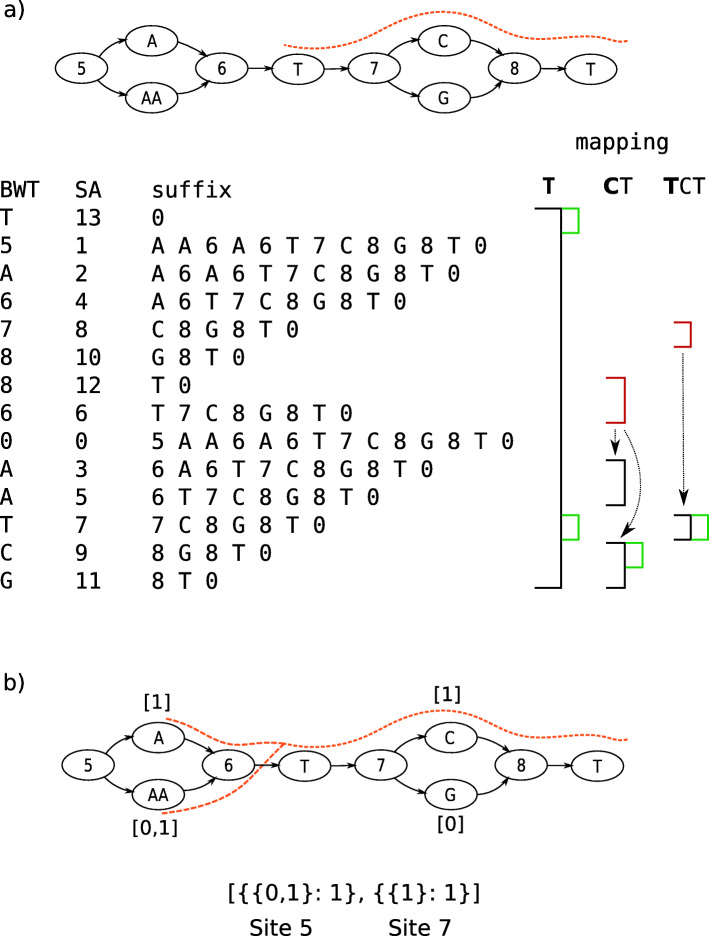
Genome graphs allow very general representations of genetic variation; depending on the model and implementation, variation at different length-scales (single nucleotide polymorphisms (SNPs), structural variants) and on different sequence backgrounds can be incorporated with different levels of transparency. We implement a model which handles this multiscale variation and develop a JSON extension of VCF (jVCF) allowing for variant calls on multiple references, both implemented in our software gramtools. We find gramtools outperforms existing methods for genotyping SNPs overlapping large deletions in M. tuberculosis and is able to genotype on multiple alternate backgrounds in P. falciparum, revealing previously hidden recombination.
Keywords: Genome graph; Mycobacterium tuberculosis; Pangenome; Plasmodium falciparum; VCF; Variant calling.
© 2021. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

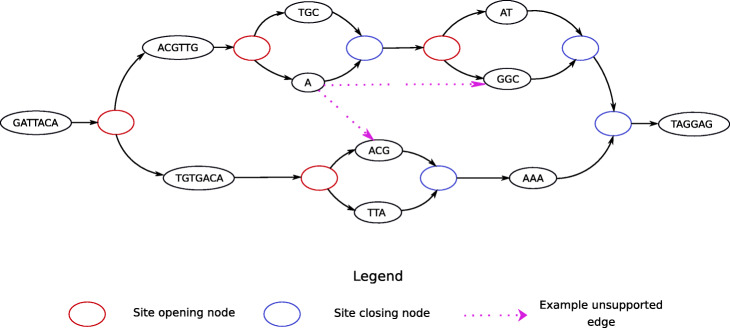

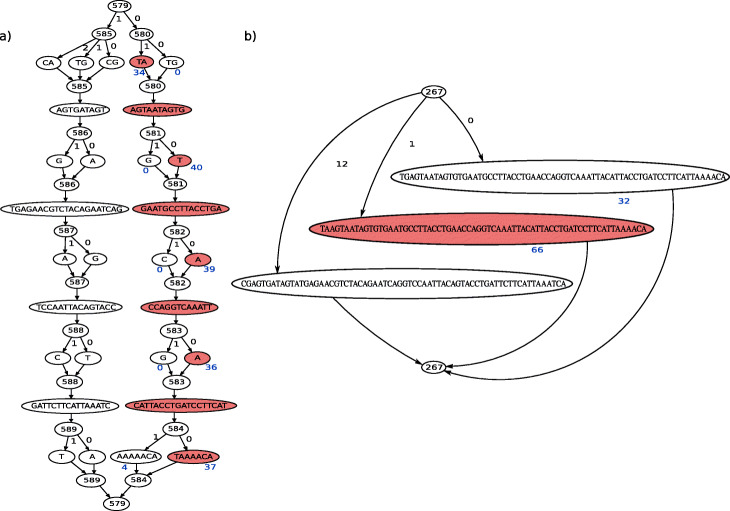

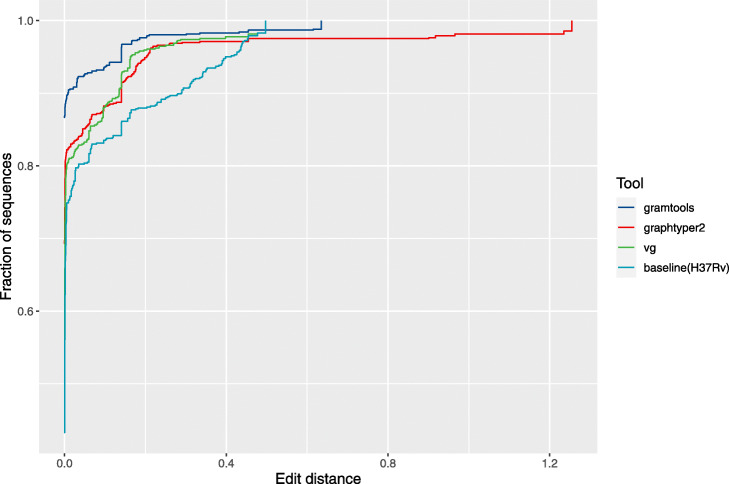
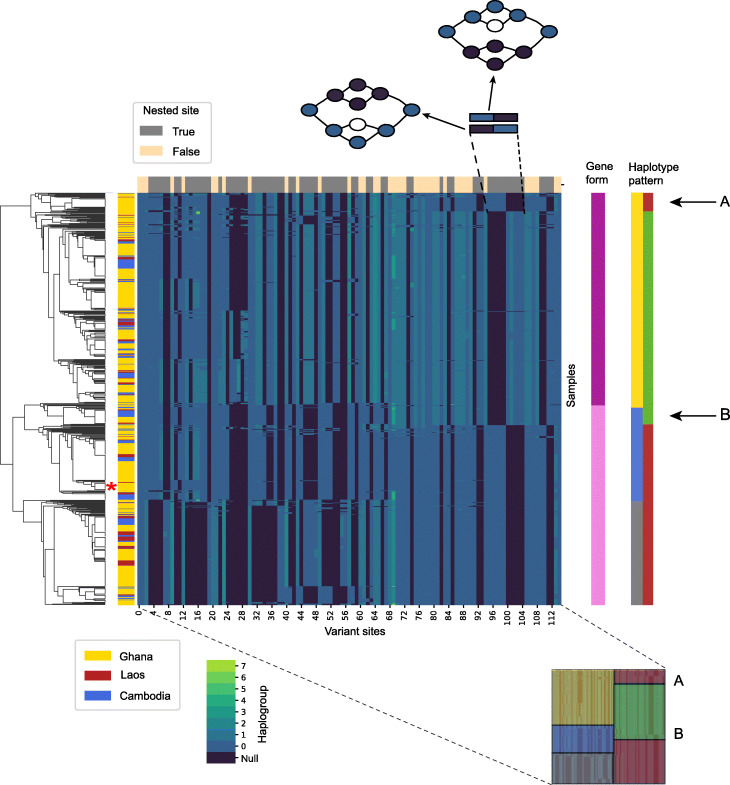

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
