

Integration of spatial and single-cell transcriptomic data elucidates mouse organogenesis
- PMID: 34489600
- PMCID: PMC8763645
- DOI: 10.1038/s41587-021-01006-2
Integration of spatial and single-cell transcriptomic data elucidates mouse organogenesis
Abstract
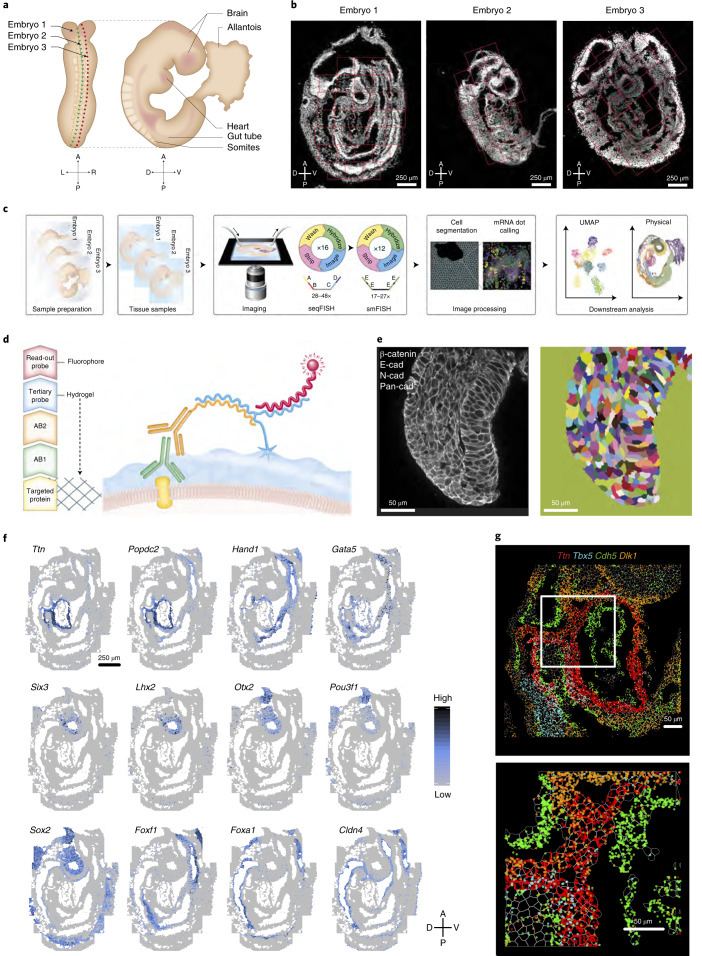
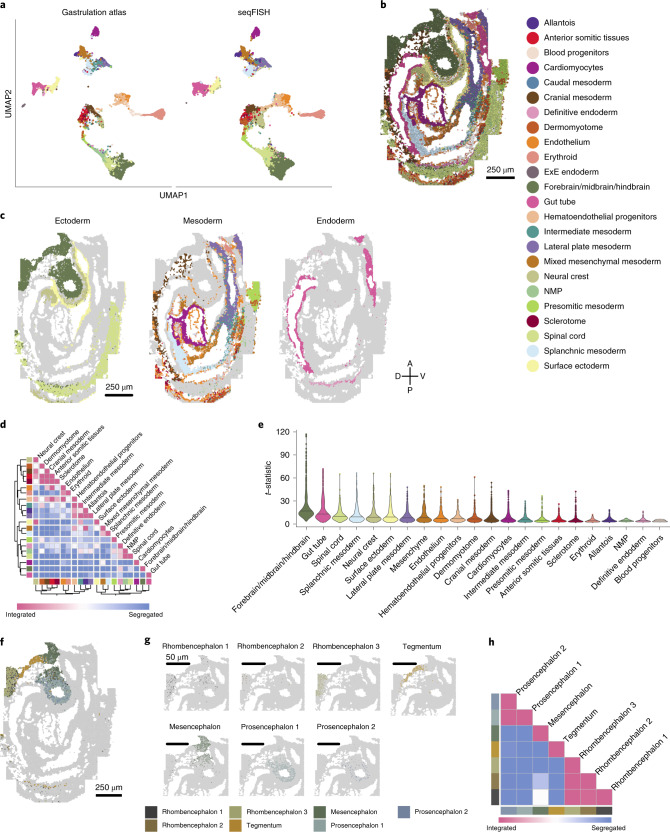
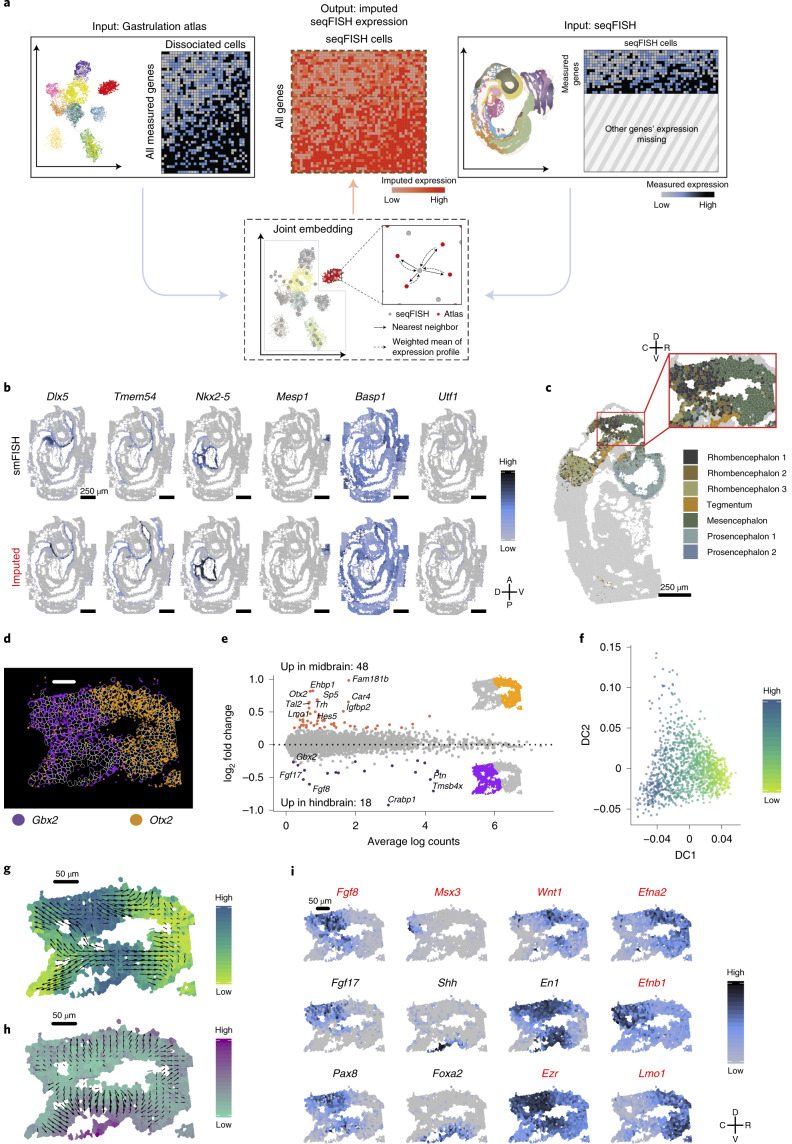
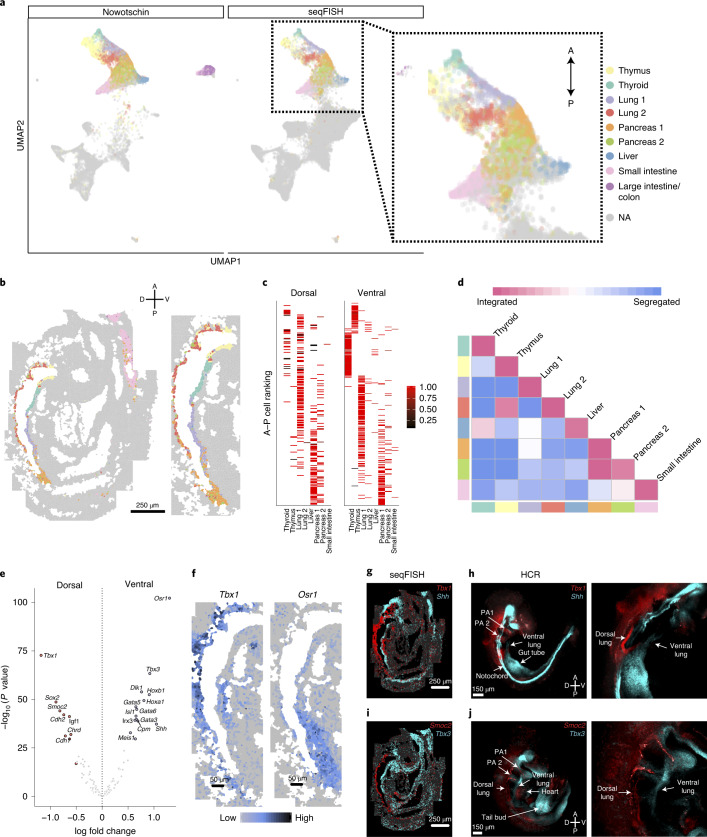
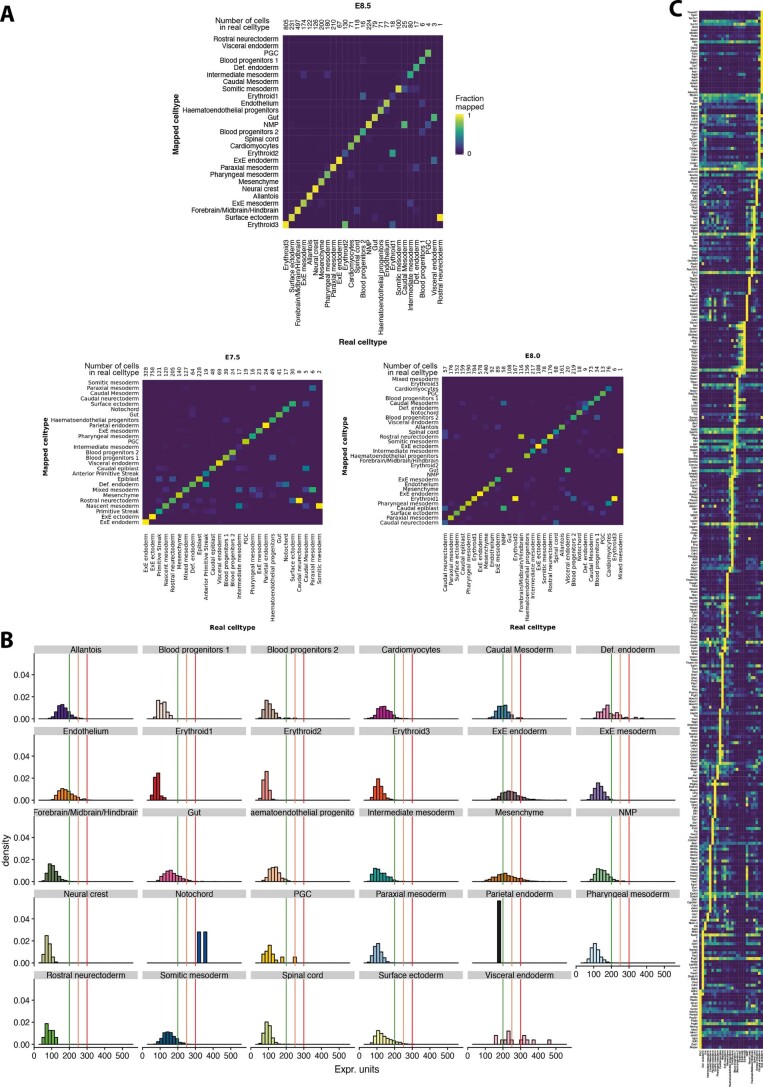
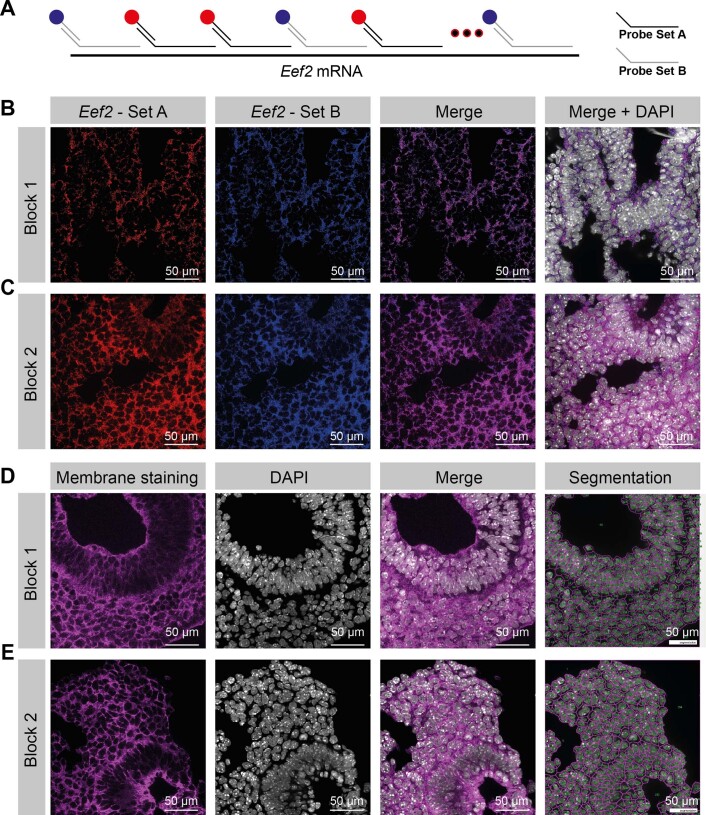
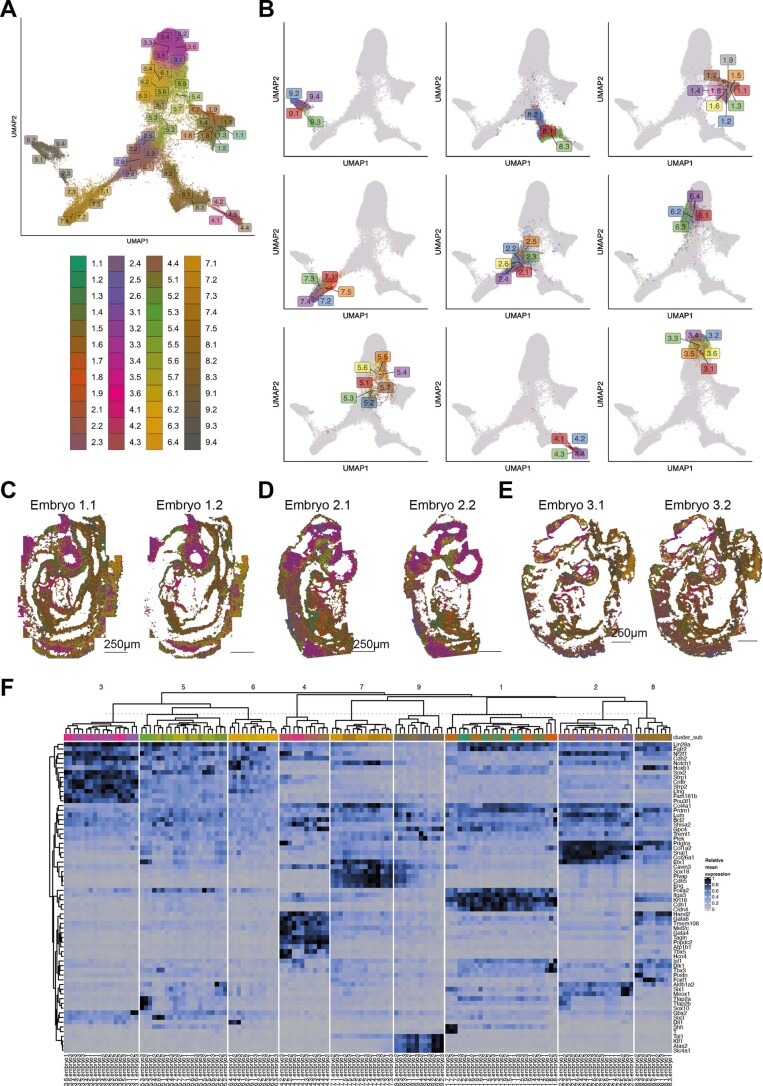
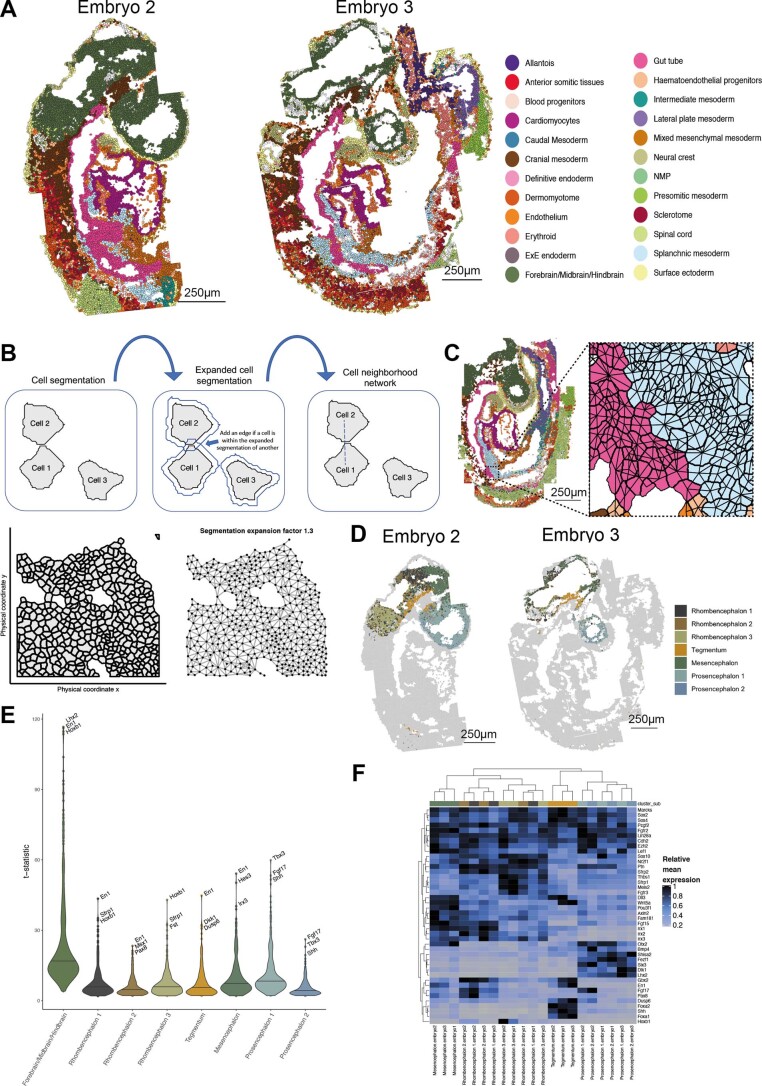
Molecular profiling of single cells has advanced our knowledge of the molecular basis of development. However, current approaches mostly rely on dissociating cells from tissues, thereby losing the crucial spatial context of regulatory processes. Here, we apply an image-based single-cell transcriptomics method, sequential fluorescence in situ hybridization (seqFISH), to detect mRNAs for 387 target genes in tissue sections of mouse embryos at the 8-12 somite stage. By integrating spatial context and multiplexed transcriptional measurements with two single-cell transcriptome atlases, we characterize cell types across the embryo and demonstrate that spatially resolved expression of genes not profiled by seqFISH can be imputed. We use this high-resolution spatial map to characterize fundamental steps in the patterning of the midbrain-hindbrain boundary (MHB) and the developing gut tube. We uncover axes of cell differentiation that are not apparent from single-cell RNA-sequencing (scRNA-seq) data, such as early dorsal-ventral separation of esophageal and tracheal progenitor populations in the gut tube. Our method provides an approach for studying cell fate decisions in complex tissues and development.
© 2021. The Author(s).
Conflict of interest statement
W.R. is a consultant and shareholder of Cambridge Epigenetix. L.C. is the cofounder of Spatial Genomics Inc. and holds patents on seqFISH. The remaining authors declare no competing interests.
Figures

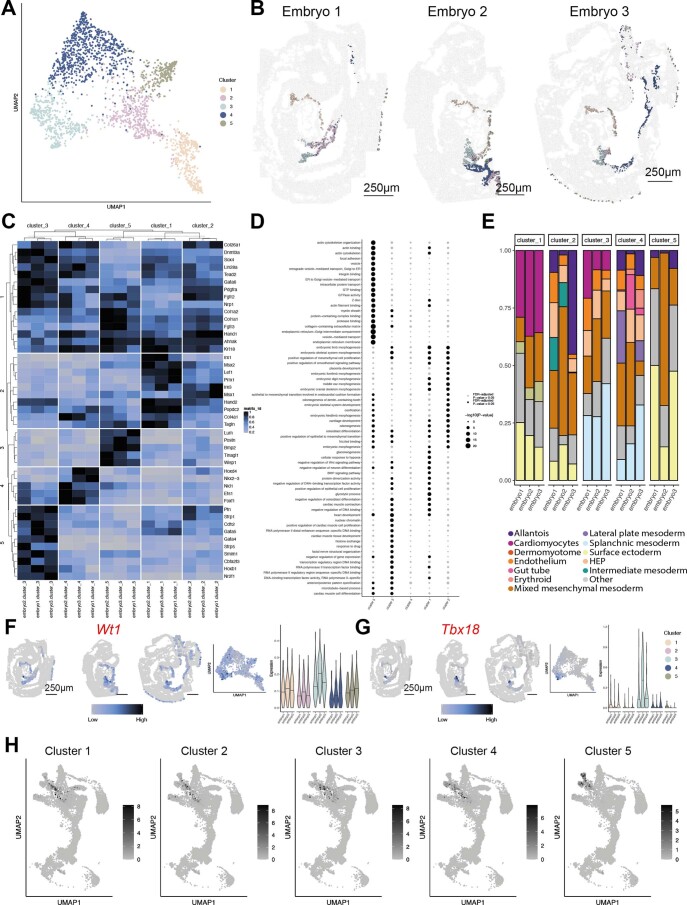
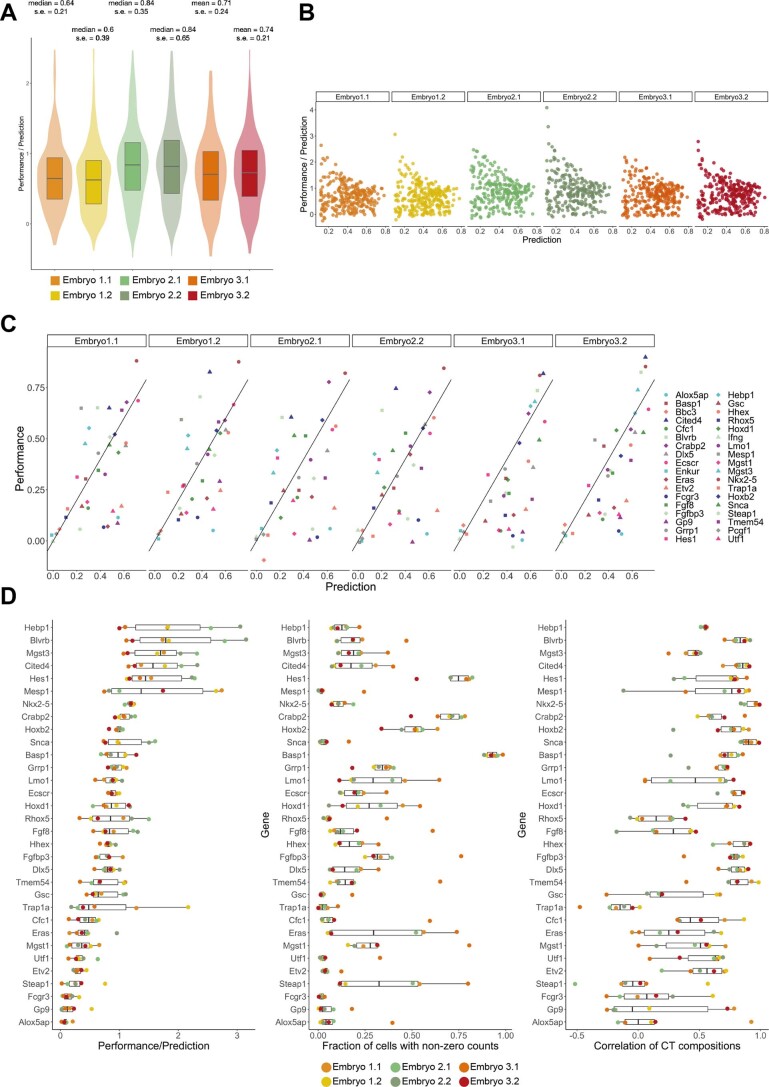

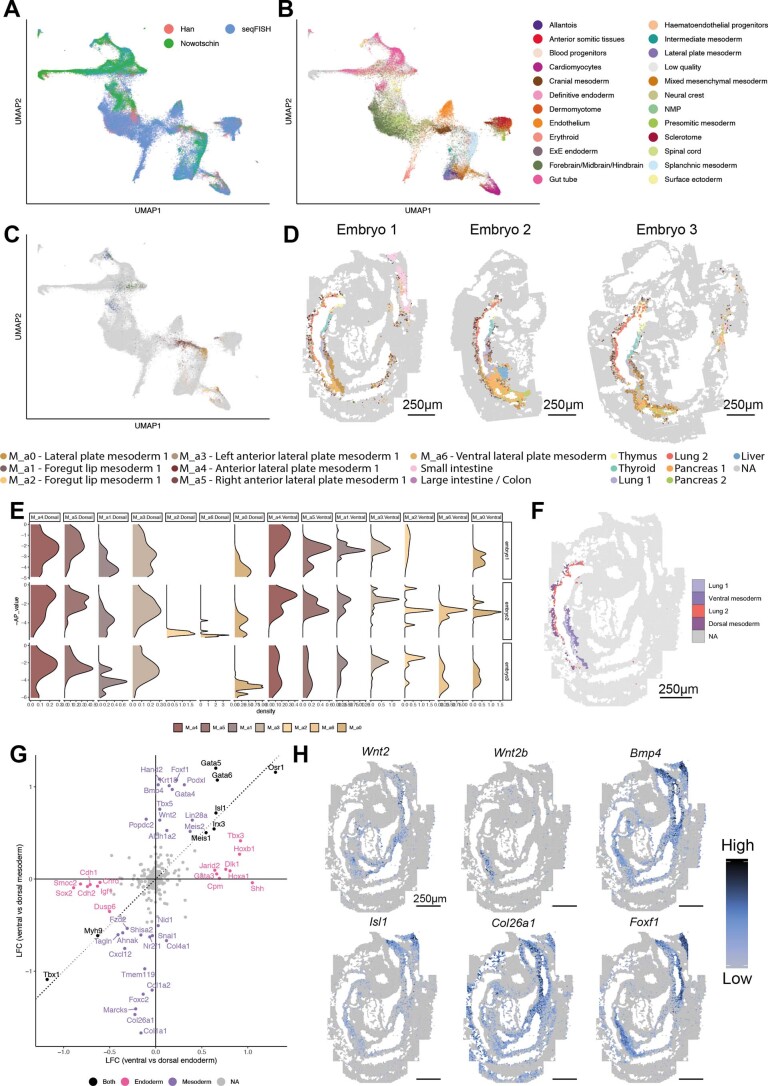
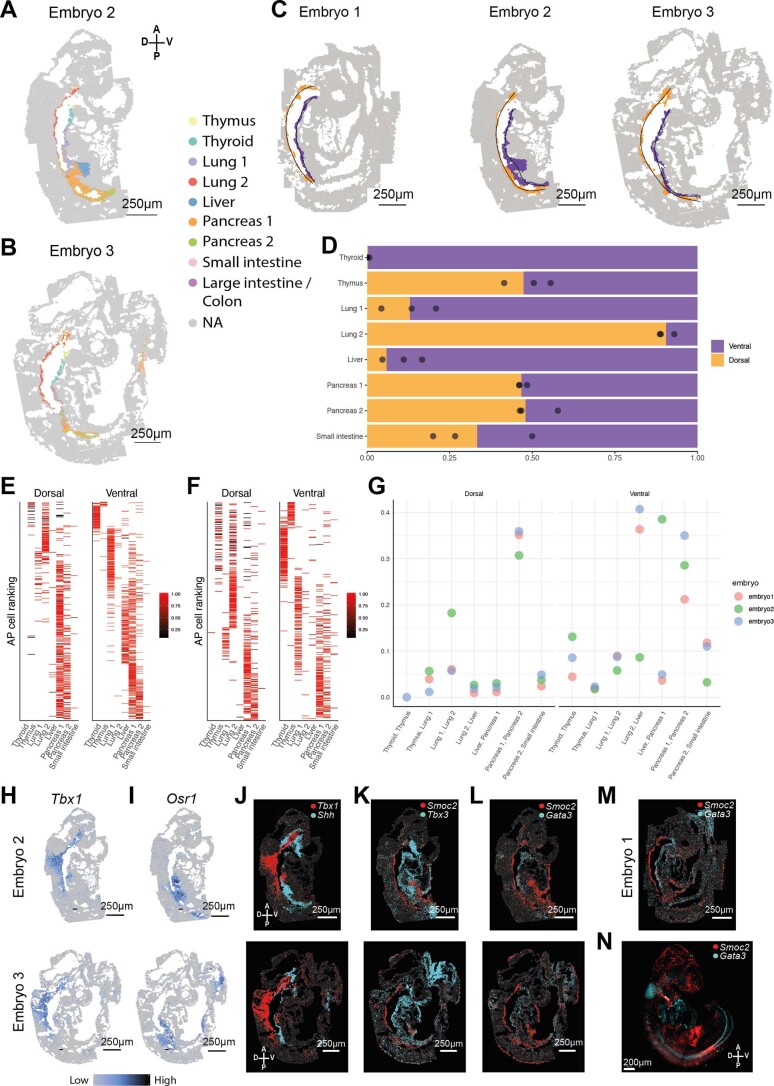
References
Publication types
MeSH terms
Substances
Grants and funding
- MC_PC_17230/MRC_/Medical Research Council/United Kingdom
- 210754/WT_/Wellcome Trust/United Kingdom
- R01 DK127821/DK/NIDDK NIH HHS/United States
- FC001051/WT_/Wellcome Trust/United Kingdom
- 105031/D/14/Z/WT_/Wellcome Trust/United Kingdom
- MR/M008975/1/MRC_/Medical Research Council/United Kingdom
- 203813/Z/16/A/WT_/Wellcome Trust/United Kingdom
- 219478/WT_/Wellcome Trust/United Kingdom
- C9545/A29580/CRUK_/Cancer Research UK/United Kingdom
- FS/18/24/33424/BHF_/British Heart Foundation/United Kingdom
- BBS/E/B/000C0421/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- P30 CA008748/CA/NCI NIH HHS/United States
- 105031/WT_/Wellcome Trust/United Kingdom
- 203813/WT_/Wellcome Trust/United Kingdom
- FC001051/CRUK_/Cancer Research UK/United Kingdom
- OT2 OD026673/OD/NIH HHS/United States
- R01 DK084391/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
