

Interpretable, Scalable, and Transferrable Functional Projection of Large-Scale Transcriptome Data Using Constrained Matrix Decomposition
- PMID: 34490045
- PMCID: PMC8417714
- DOI: 10.3389/fgene.2021.719099
Interpretable, Scalable, and Transferrable Functional Projection of Large-Scale Transcriptome Data Using Constrained Matrix Decomposition
Abstract
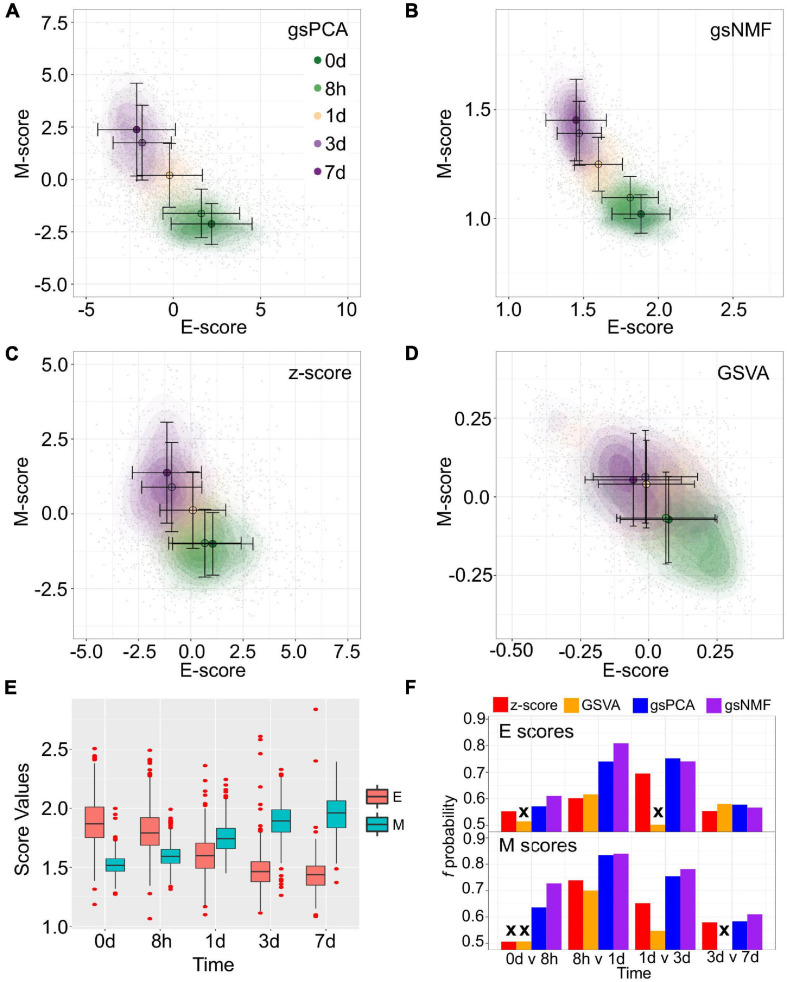
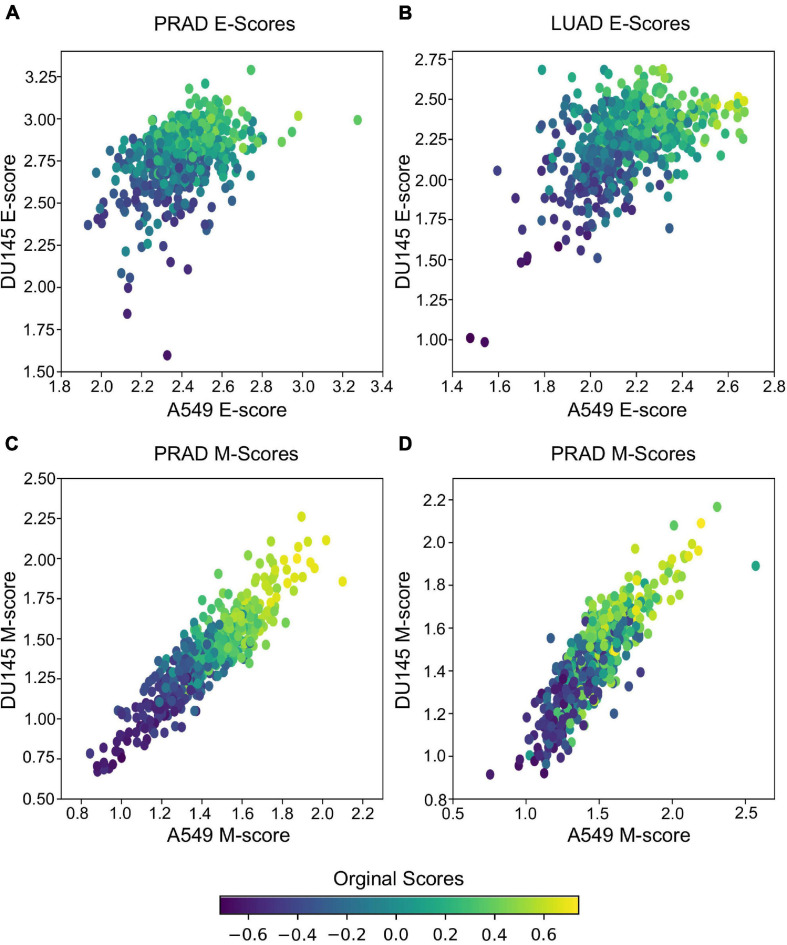
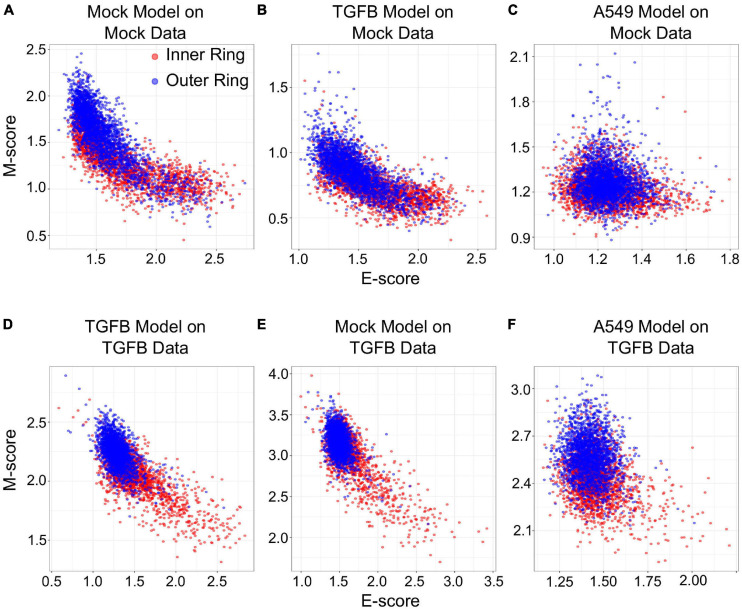
Large-scale transcriptome data, such as single-cell RNA-sequencing data, have provided unprecedented resources for studying biological processes at the systems level. Numerous dimensionality reduction methods have been developed to visualize and analyze these transcriptome data. In addition, several existing methods allow inference of functional variations among samples using gene sets with known biological functions. However, it remains challenging to analyze transcriptomes with reduced dimensions that are interpretable in terms of dimensions' directionalities, transferrable to new data, and directly expose the contribution or association of individual genes. In this study, we used gene set non-negative principal component analysis (gsPCA) and non-negative matrix factorization (gsNMF) to analyze large-scale transcriptome datasets. We found that these methods provide low-dimensional information about the progression of biological processes in a quantitative manner, and their performances are comparable to existing functional variation analysis methods in terms of distinguishing multiple cell states and samples from multiple conditions. Remarkably, upon training with a subset of data, these methods allow predictions of locations in the functional space using data from experimental conditions that are not exposed to the models. Specifically, our models predicted the extent of progression and reversion for cells in the epithelial-mesenchymal transition (EMT) continuum. These methods revealed conserved EMT program among multiple types of single cells and tumor samples. Finally, we demonstrate this approach is broadly applicable to data and gene sets beyond EMT and provide several recommendations on the choice between the two linear methods and the optimal algorithmic parameters. Our methods show that simple constrained matrix decomposition can produce to low-dimensional information in functionally interpretable and transferrable space, and can be widely useful for analyzing large-scale transcriptome data.
Keywords: EMT; RNA-sequencing data; dimensionality reduction; gene set analysis; single-cell ‘omics.
Copyright © 2021 Panchy, Watanabe and Hong.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures





References
Grants and funding
LinkOut - more resources
Full Text Sources