

Extracellular Vesicles in Sickle Cell Disease: Plasma Concentration, Blood Cell Types Origin Distribution and Biological Properties
- PMID: 34490315
- PMCID: PMC8417591
- DOI: 10.3389/fmed.2021.728693
Extracellular Vesicles in Sickle Cell Disease: Plasma Concentration, Blood Cell Types Origin Distribution and Biological Properties
Abstract
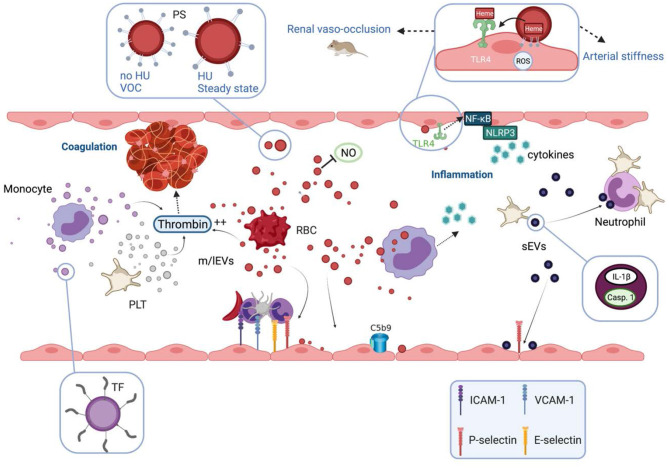
Prototype of monogenic disorder, sickle cell disease (SCD) is caused by a unique single mutation in the β-globin gene, leading to the production of the abnormal hemoglobin S (HbS). HbS polymerization in deoxygenated condition induces the sickling of red blood cells (RBCs), which become less deformable and more fragile, and thus prone to lysis. In addition to anemia, SCD patients may exhibit a plethora of clinical manifestations ranging from acute complications such as the frequent and debilitating painful vaso-occlusive crisis to chronic end organ damages. Several interrelated pathophysiological processes have been described, including impaired blood rheology, increased blood cell adhesion, coagulation, inflammation and enhanced oxidative stress among others. During the last two decades, it has been shown that extracellular vesicles (EVs), defined as cell-derived anucleated particles delimited by a lipid bilayer, and comprising small EVs (sEVs) and medium/large EVs (m/lEVs); are not only biomarkers but also subcellular actors in SCD pathophysiology. Plasma concentration of m/lEVs, originated mainly from RBCs and platelets (PLTs) but also from the other blood cell types, is higher in SCD patients than in healthy controls. The concentration and the density of externalized phosphatidylserine of those released from RBCs may vary according to clinical status (crisis vs. steady state) and treatment (hydroxyurea). Besides their procoagulant properties initially described, RBC-m/lEVs may promote inflammation through their effects on monocytes/macrophages and endothelial cells. Although less intensely studied, sEVs plasma concentration is increased in SCD and these EVs may cause endothelial damages. In addition, sEVs released from activated PLTs trigger PLT-neutrophil aggregation involved in lung vaso-occlusion in sickle mice. Altogether, these data clearly indicate that EVs are both biomarkers and bio-effectors in SCD, which deserve further studies.
Keywords: coagulation; endothelial dysfunction; extracellular vesicles; inflammation; oxidative stress; sickle cell disease.
Copyright © 2021 Nader, Garnier, Connes and Romana.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources