

Mass Spectrometry-Based Techniques to Elucidate the Sugar Code
- PMID: 34491038
- PMCID: PMC9052437
- DOI: 10.1021/acs.chemrev.1c00380
Mass Spectrometry-Based Techniques to Elucidate the Sugar Code
Abstract
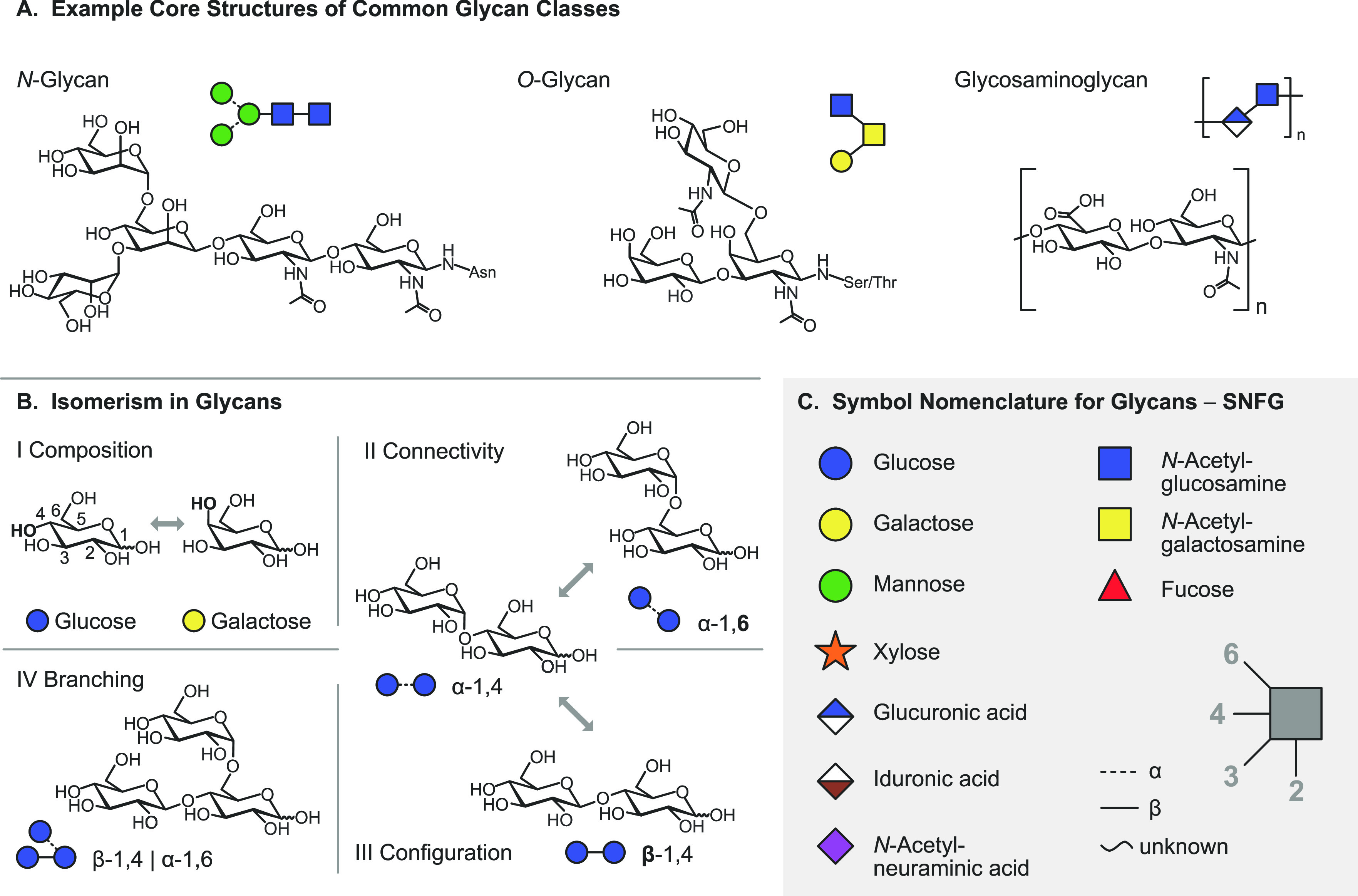
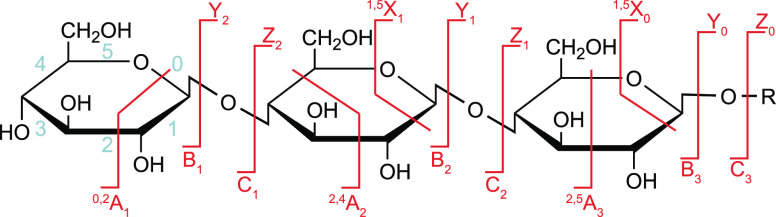
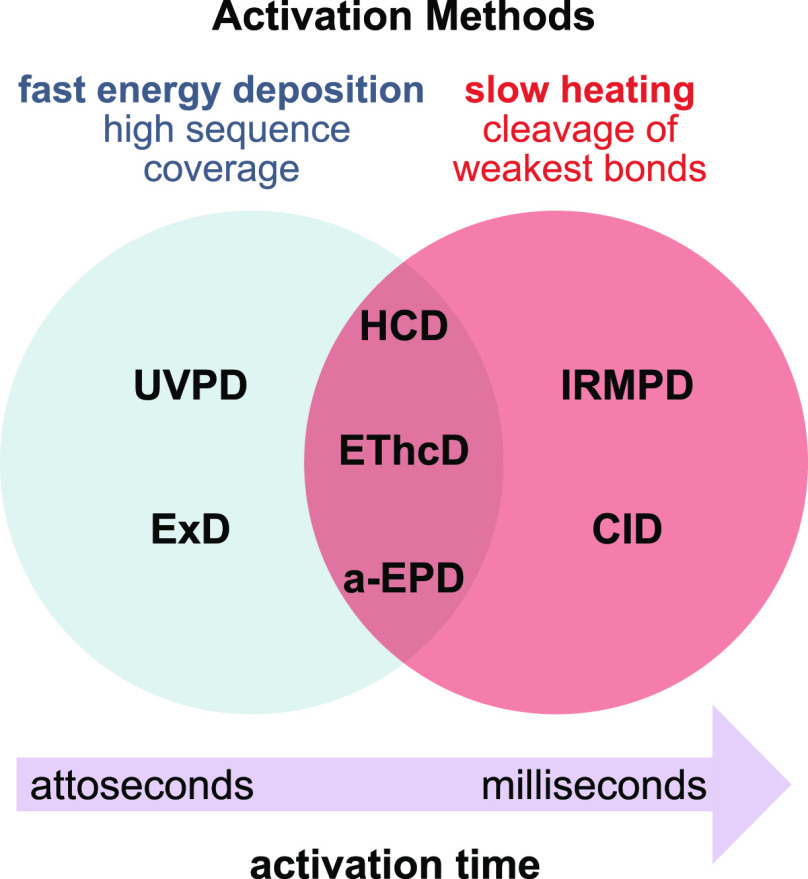
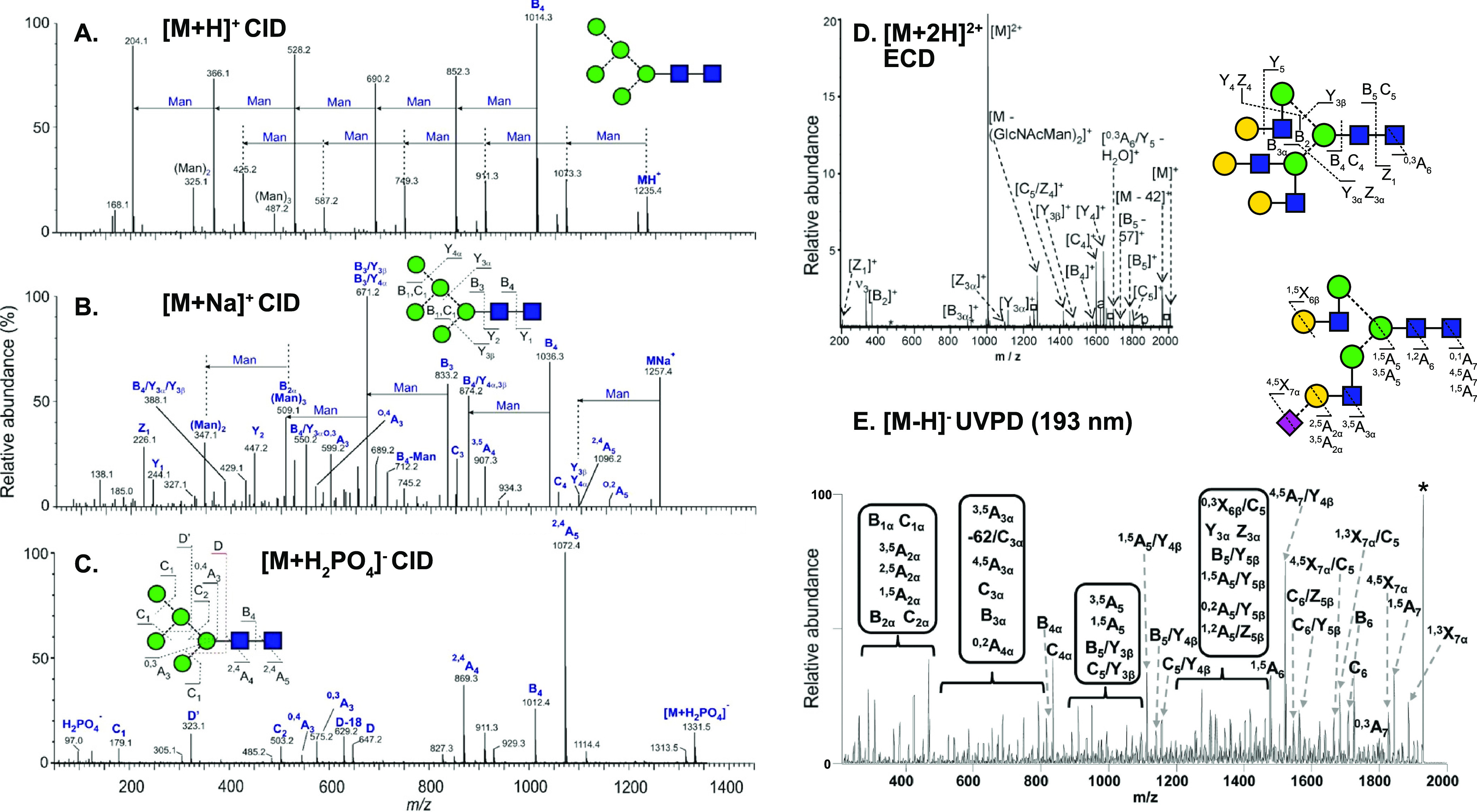
Cells encode information in the sequence of biopolymers, such as nucleic acids, proteins, and glycans. Although glycans are essential to all living organisms, surprisingly little is known about the "sugar code" and the biological roles of these molecules. The reason glycobiology lags behind its counterparts dealing with nucleic acids and proteins lies in the complexity of carbohydrate structures, which renders their analysis extremely challenging. Building blocks that may differ only in the configuration of a single stereocenter, combined with the vast possibilities to connect monosaccharide units, lead to an immense variety of isomers, which poses a formidable challenge to conventional mass spectrometry. In recent years, however, a combination of innovative ion activation methods, commercialization of ion mobility-mass spectrometry, progress in gas-phase ion spectroscopy, and advances in computational chemistry have led to a revolution in mass spectrometry-based glycan analysis. The present review focuses on the above techniques that expanded the traditional glycomics toolkit and provided spectacular insight into the structure of these fascinating biomolecules. To emphasize the specific challenges associated with them, major classes of mammalian glycans are discussed in separate sections. By doing so, we aim to put the spotlight on the most important element of glycobiology: the glycans themselves.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
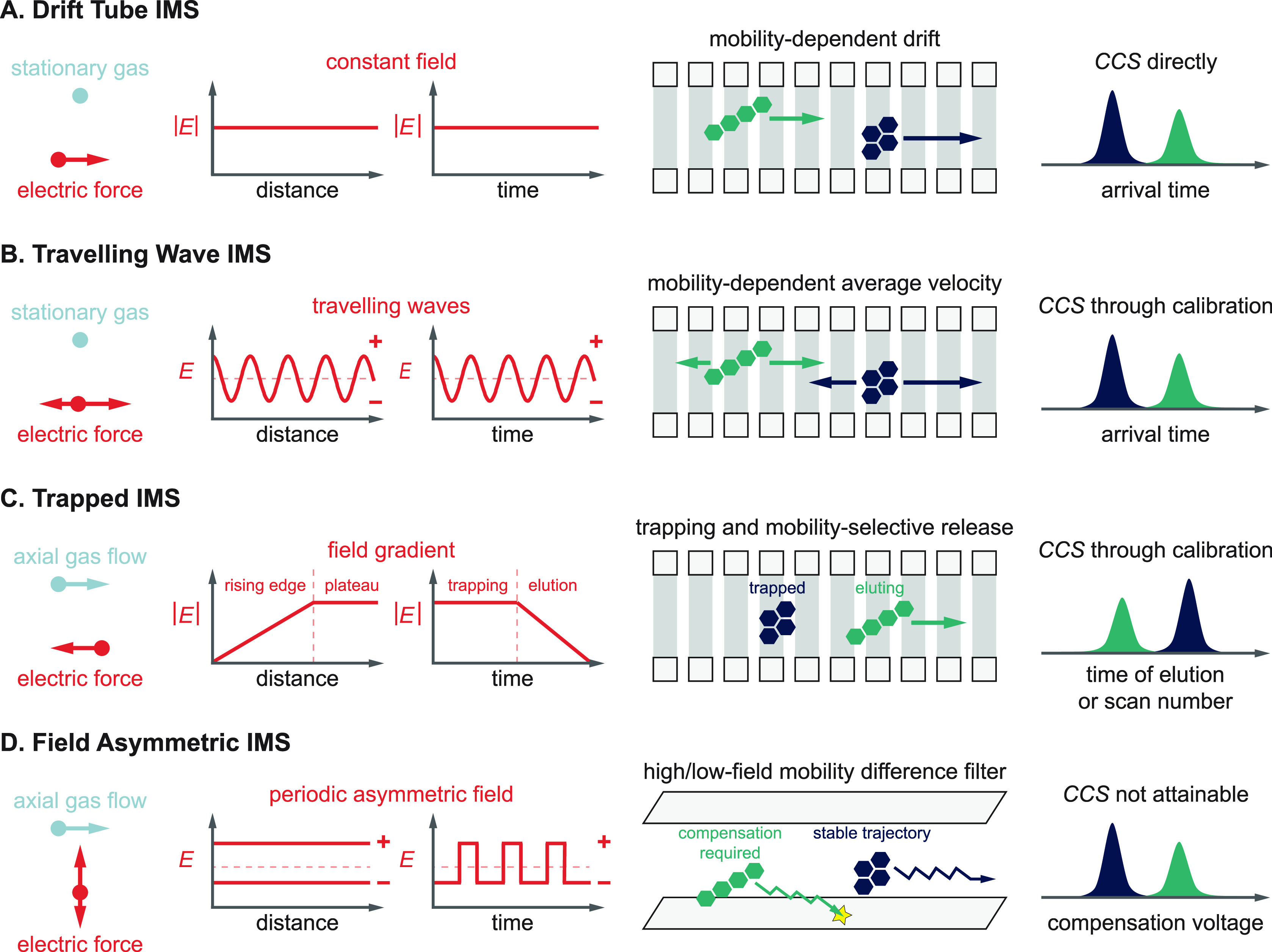
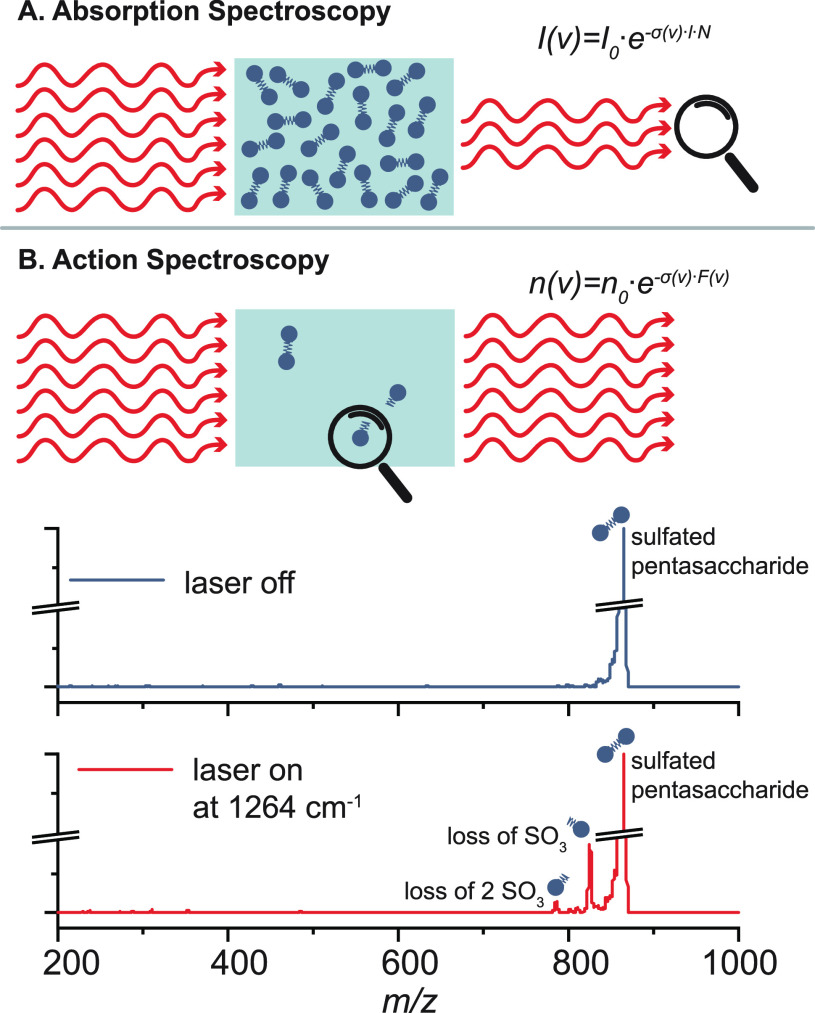
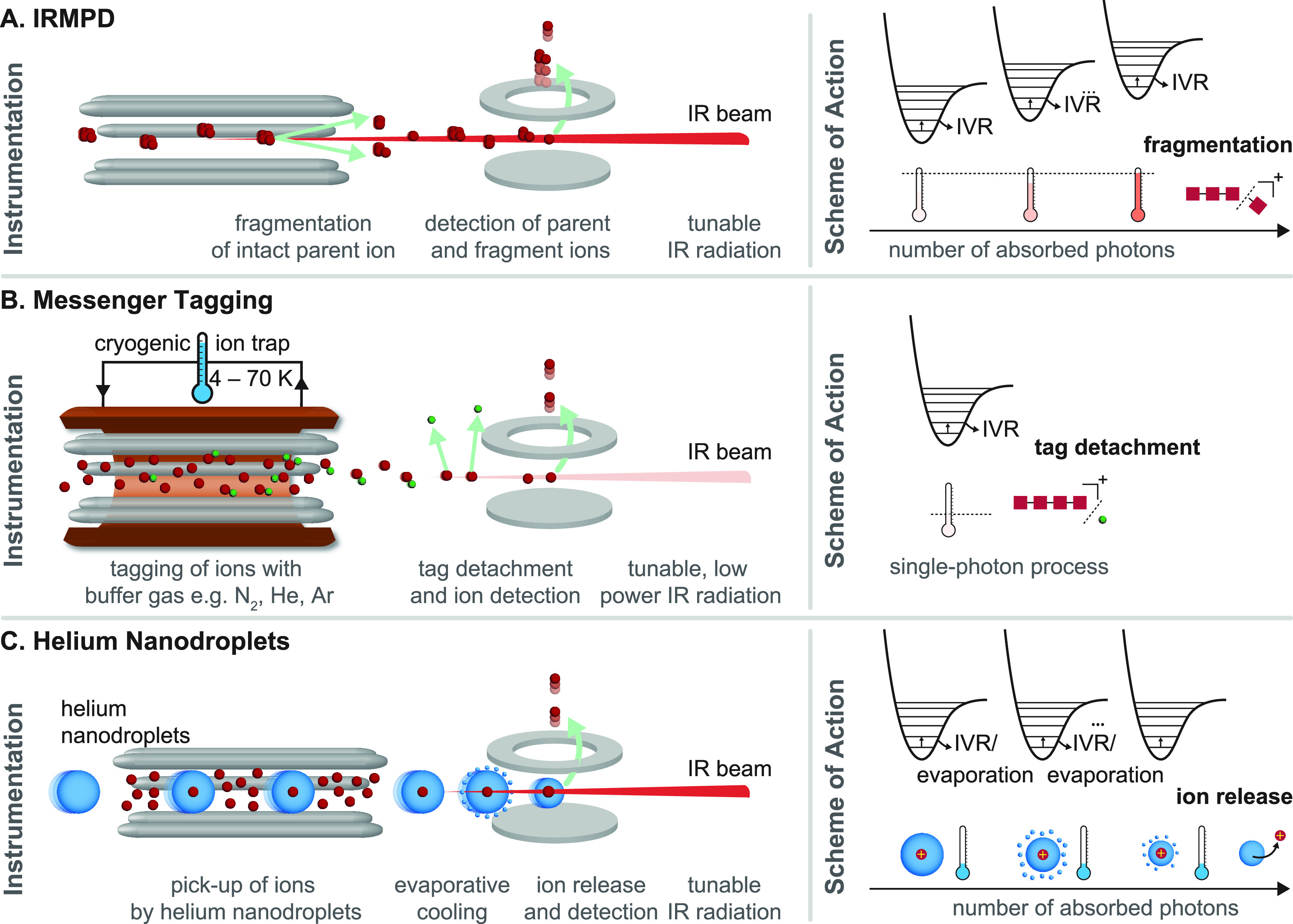
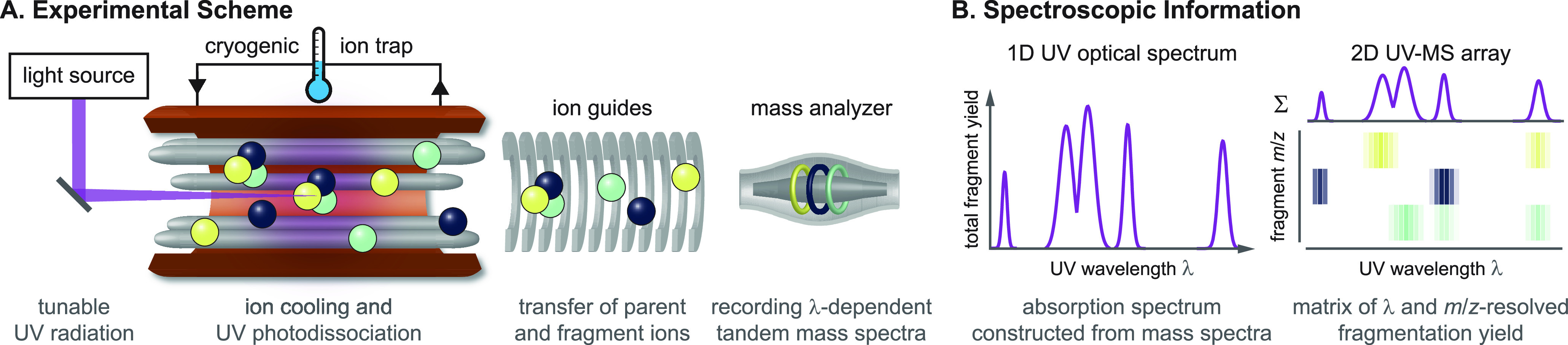
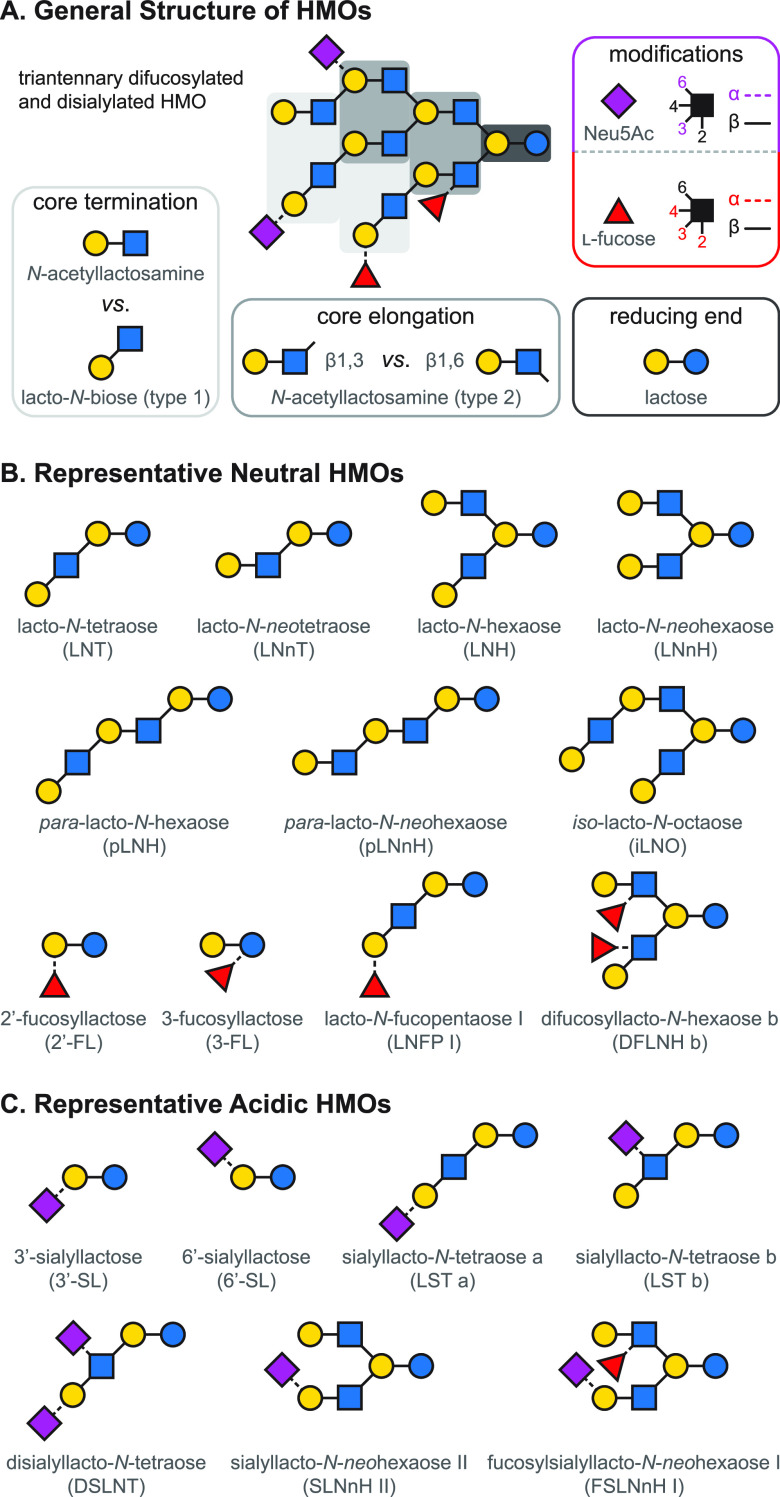
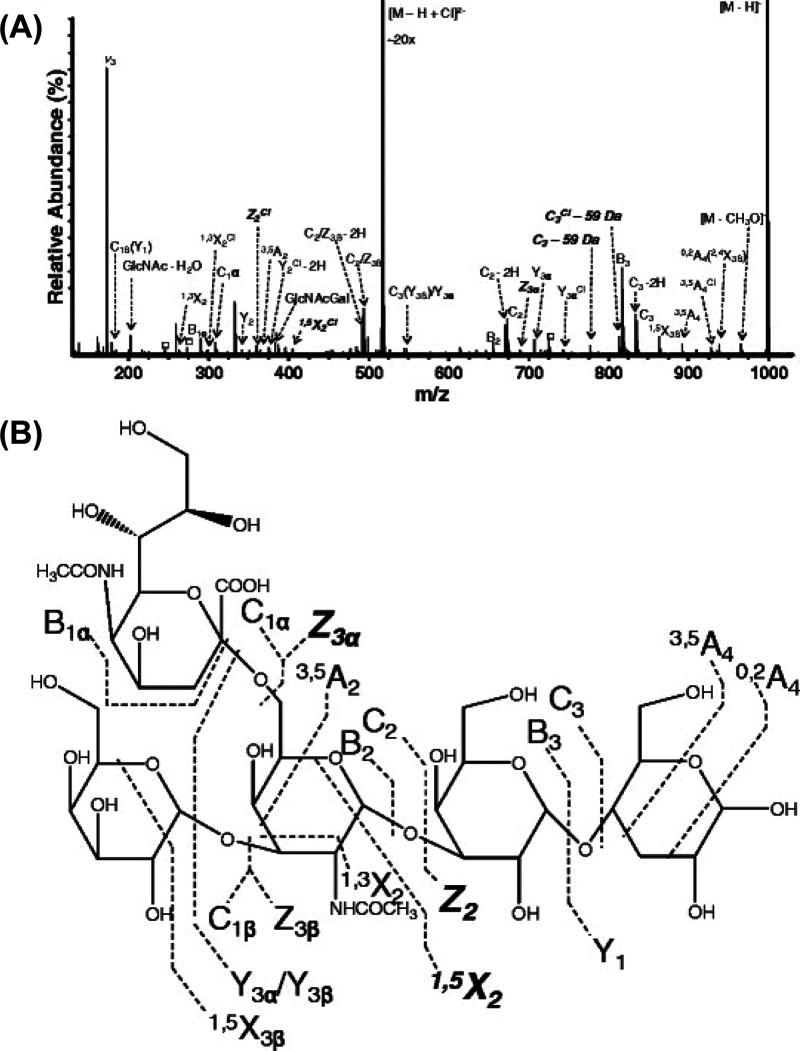
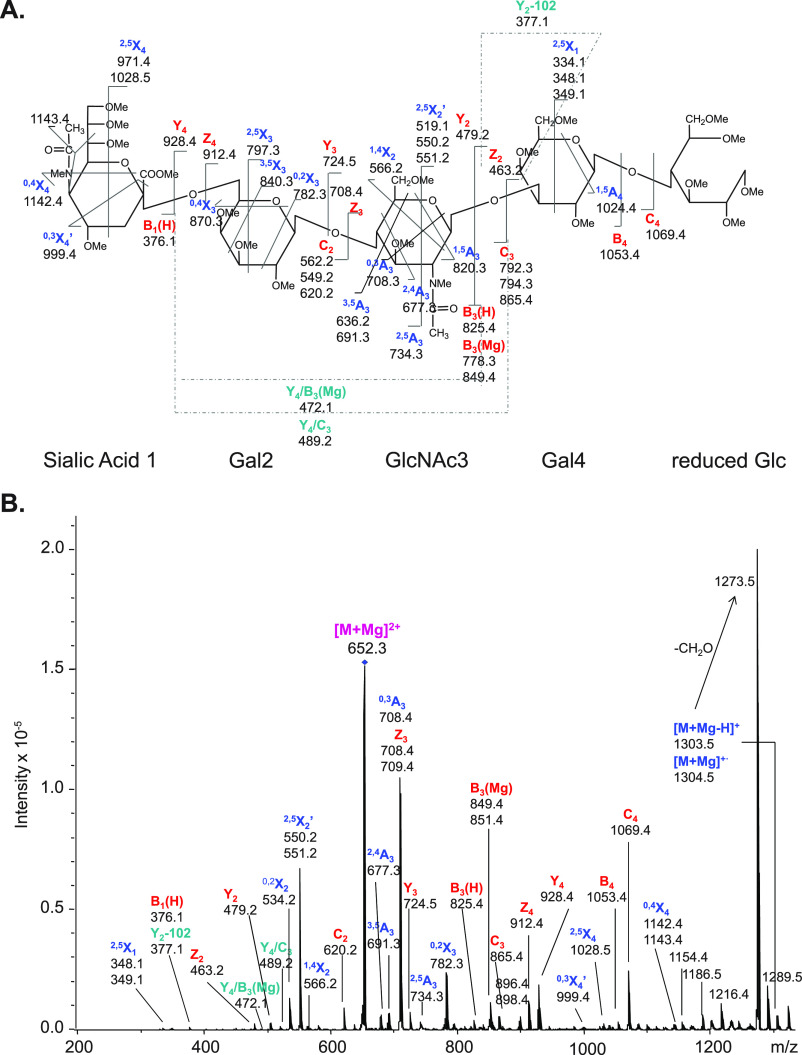
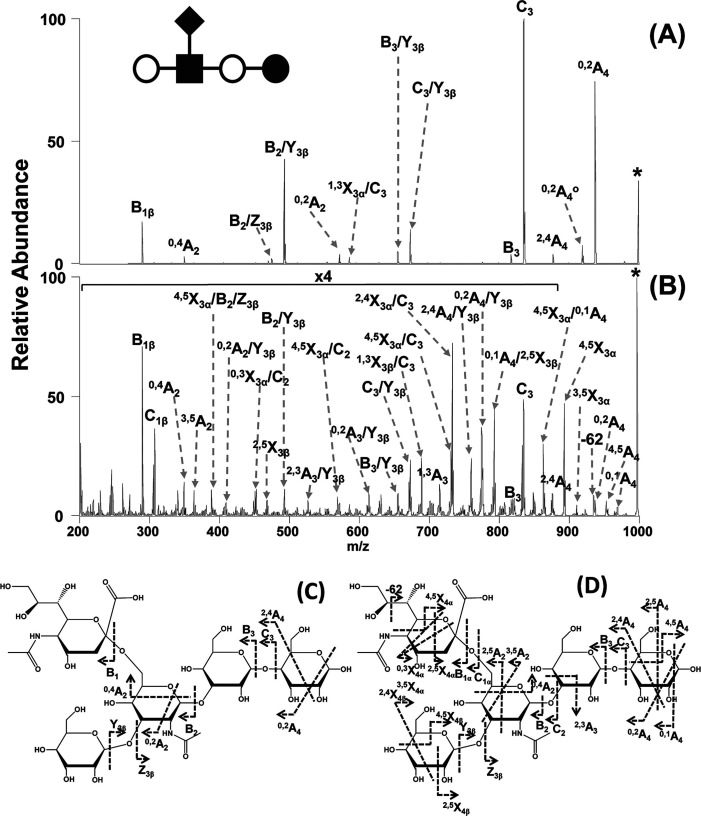
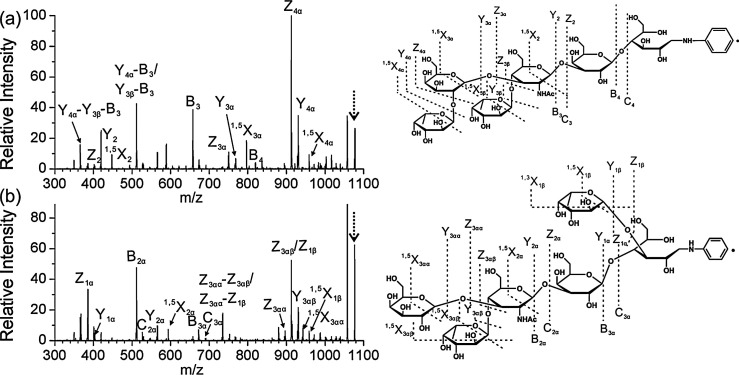
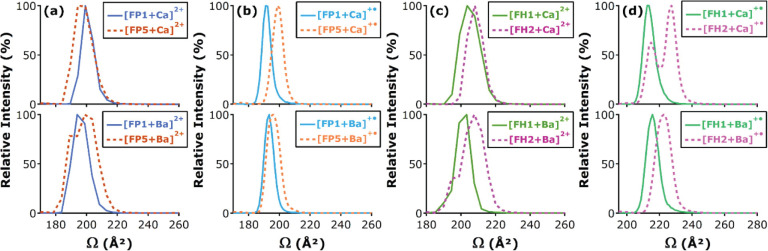
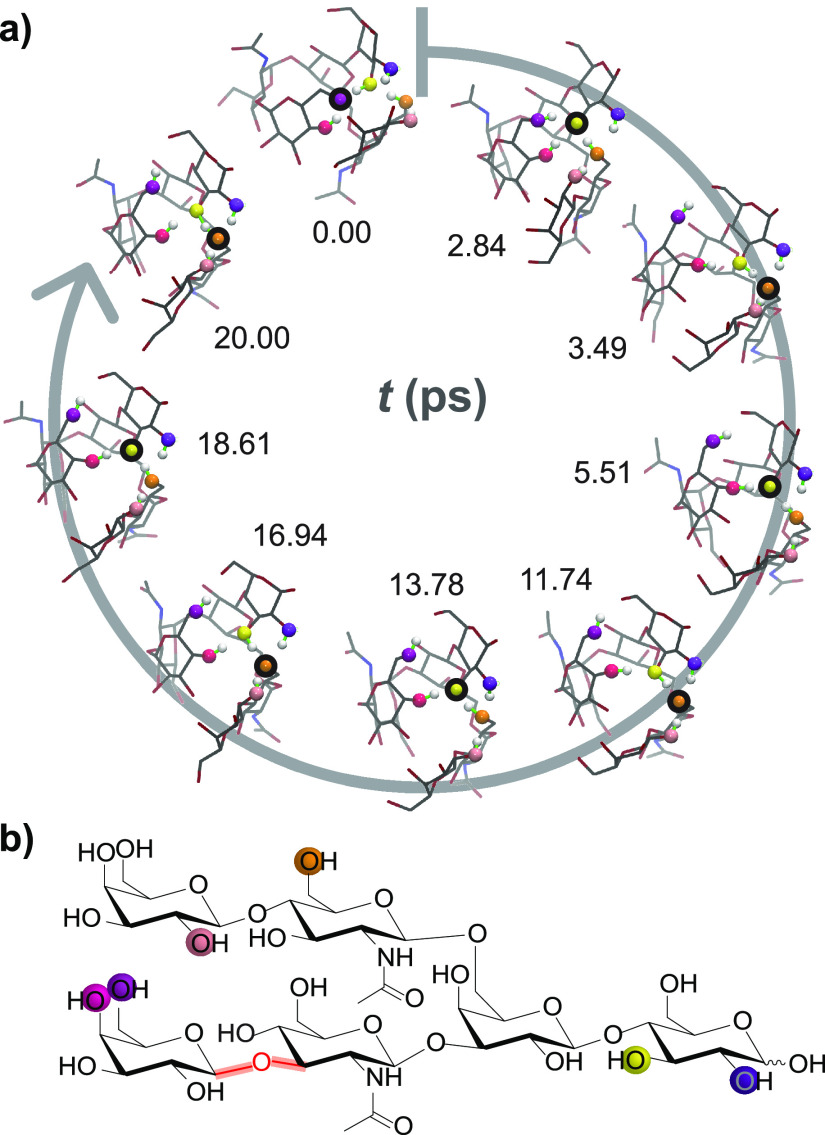
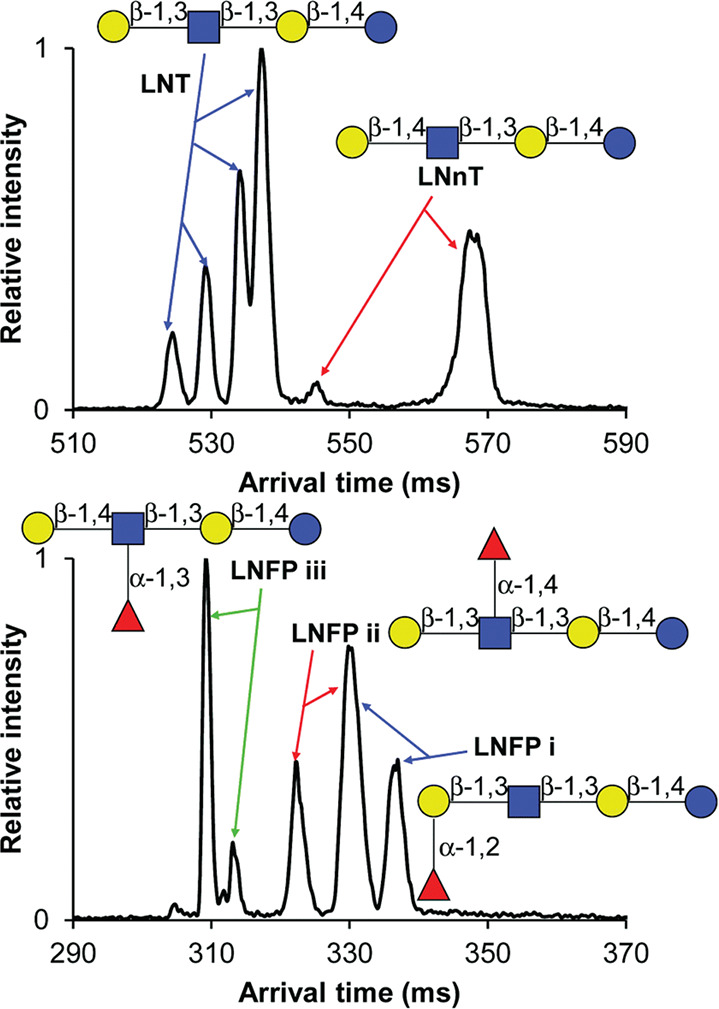
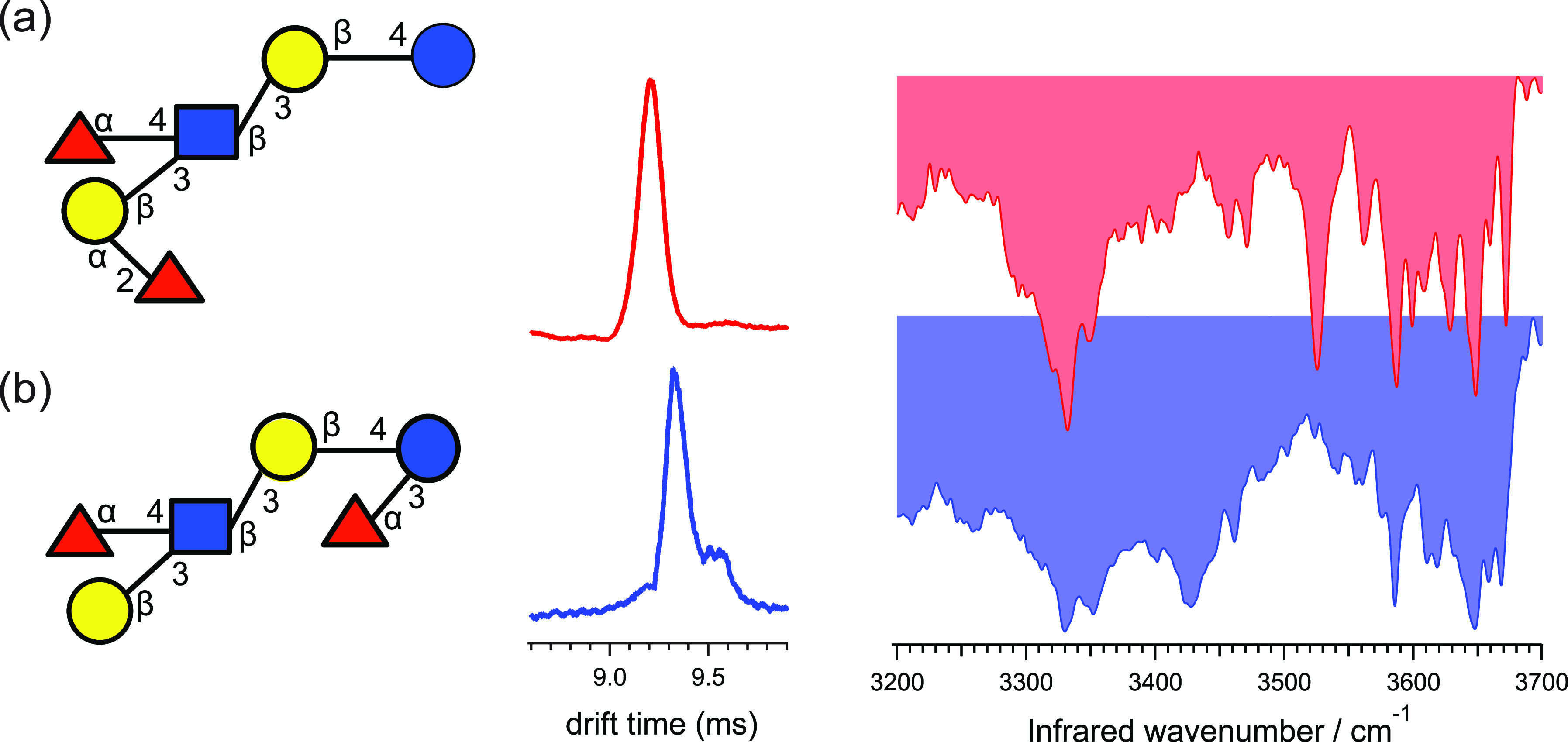
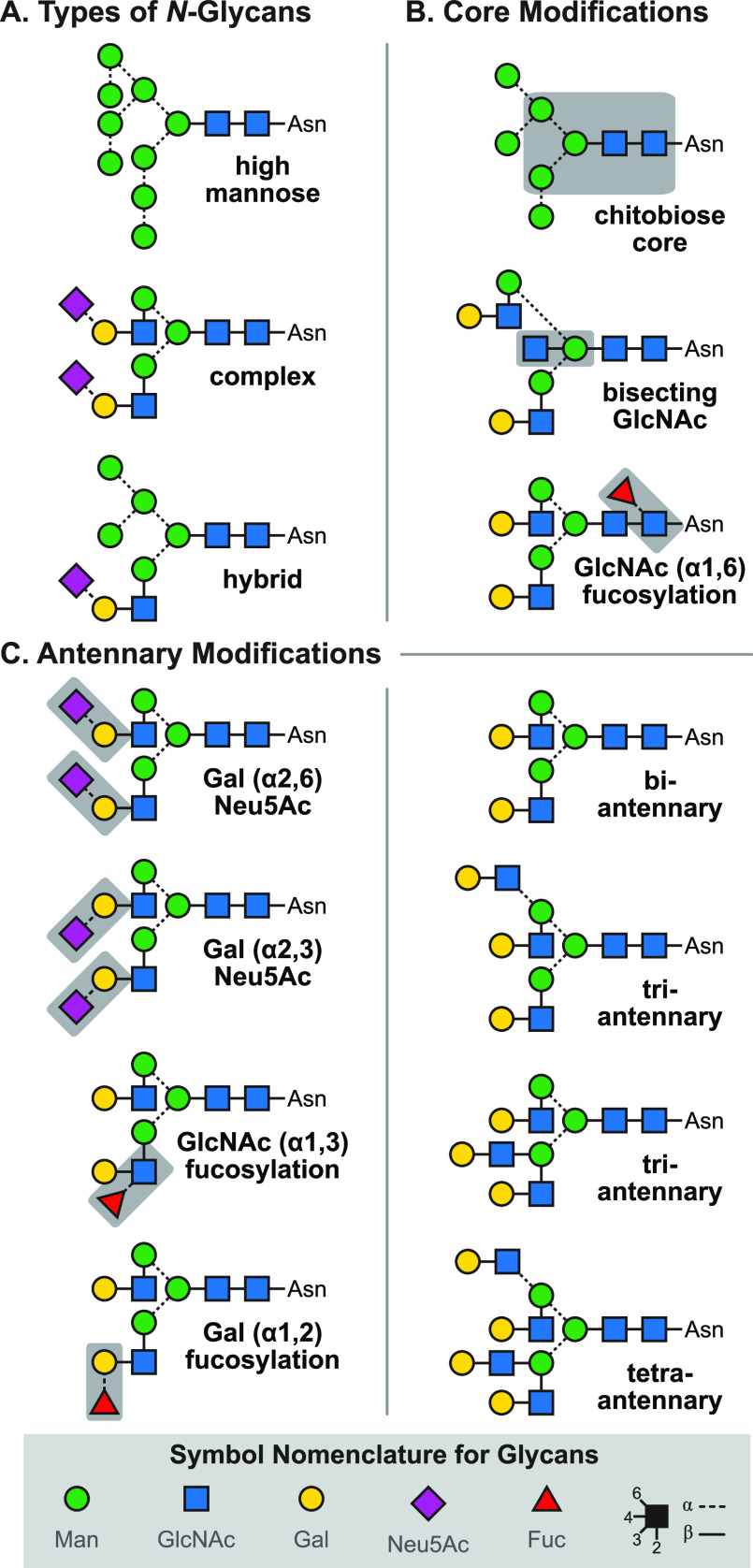
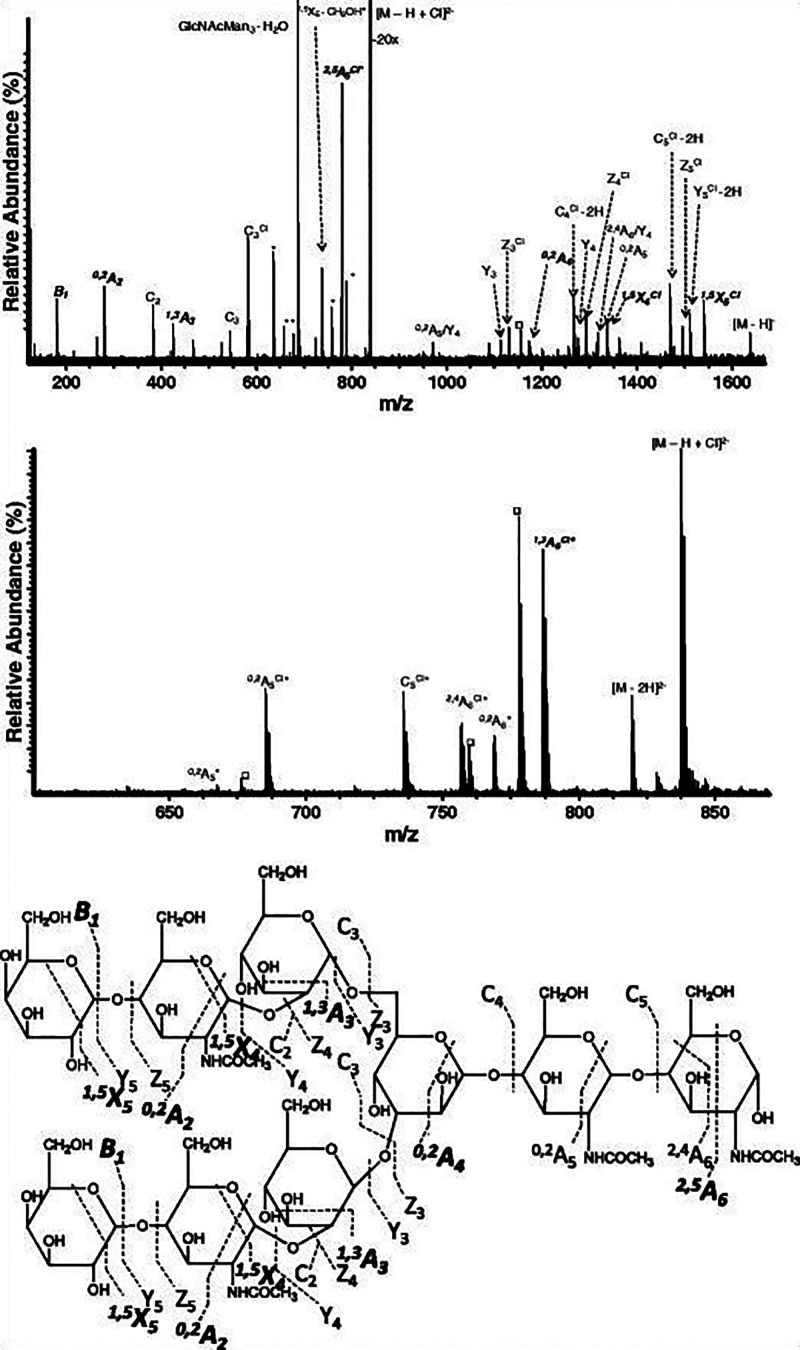
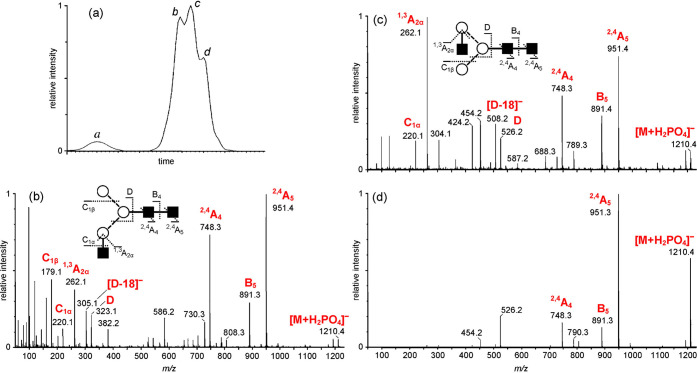
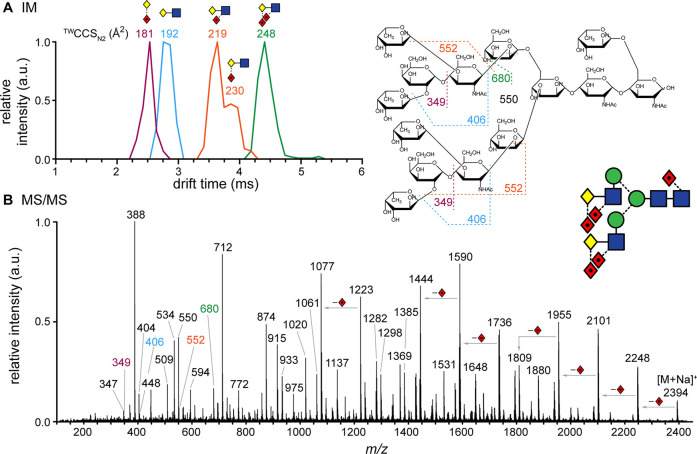
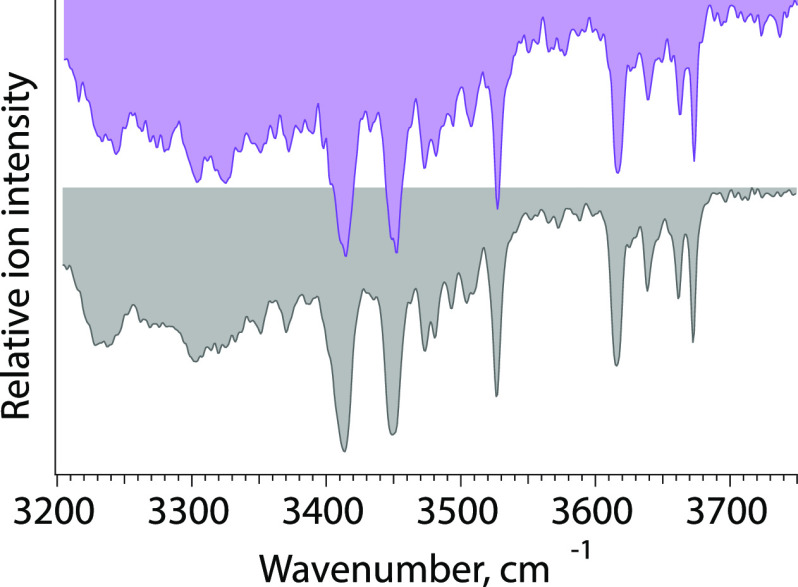
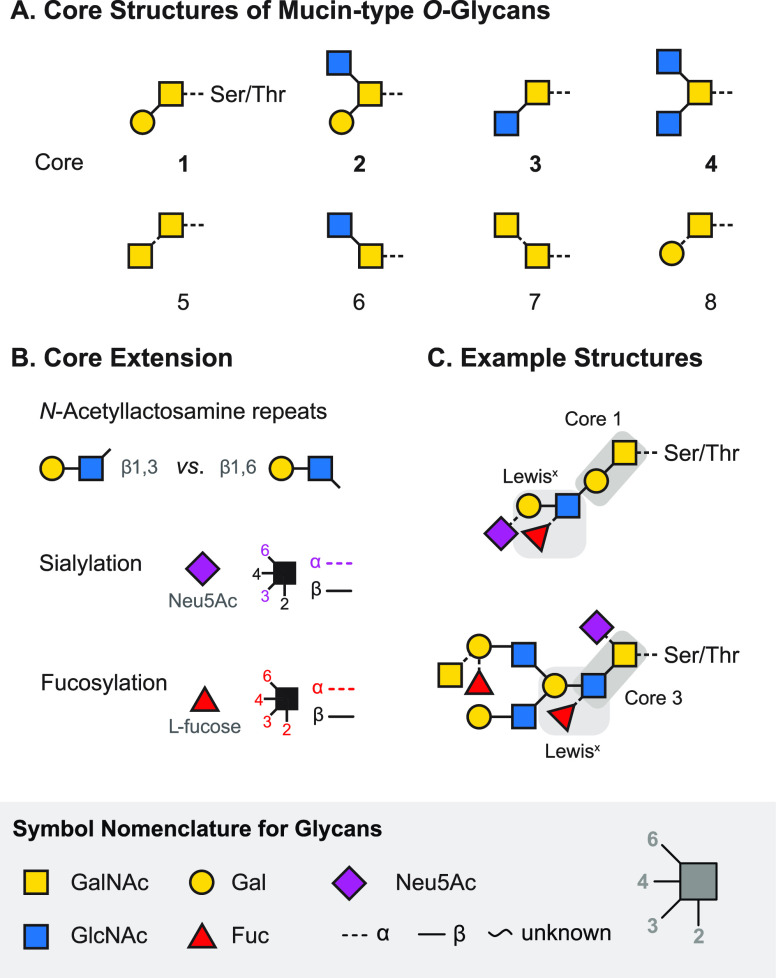
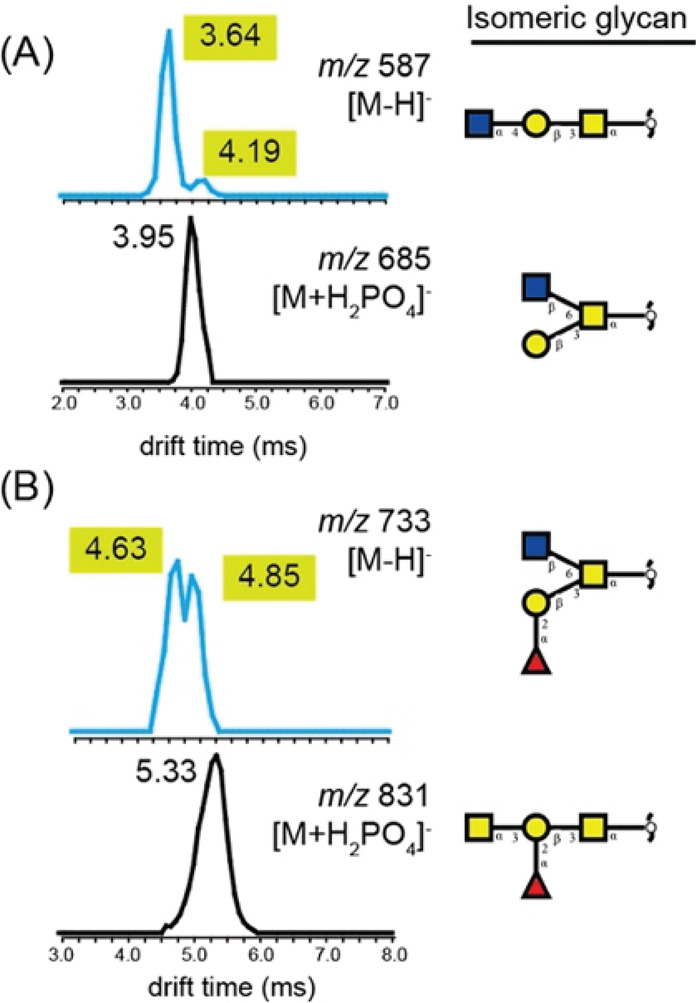
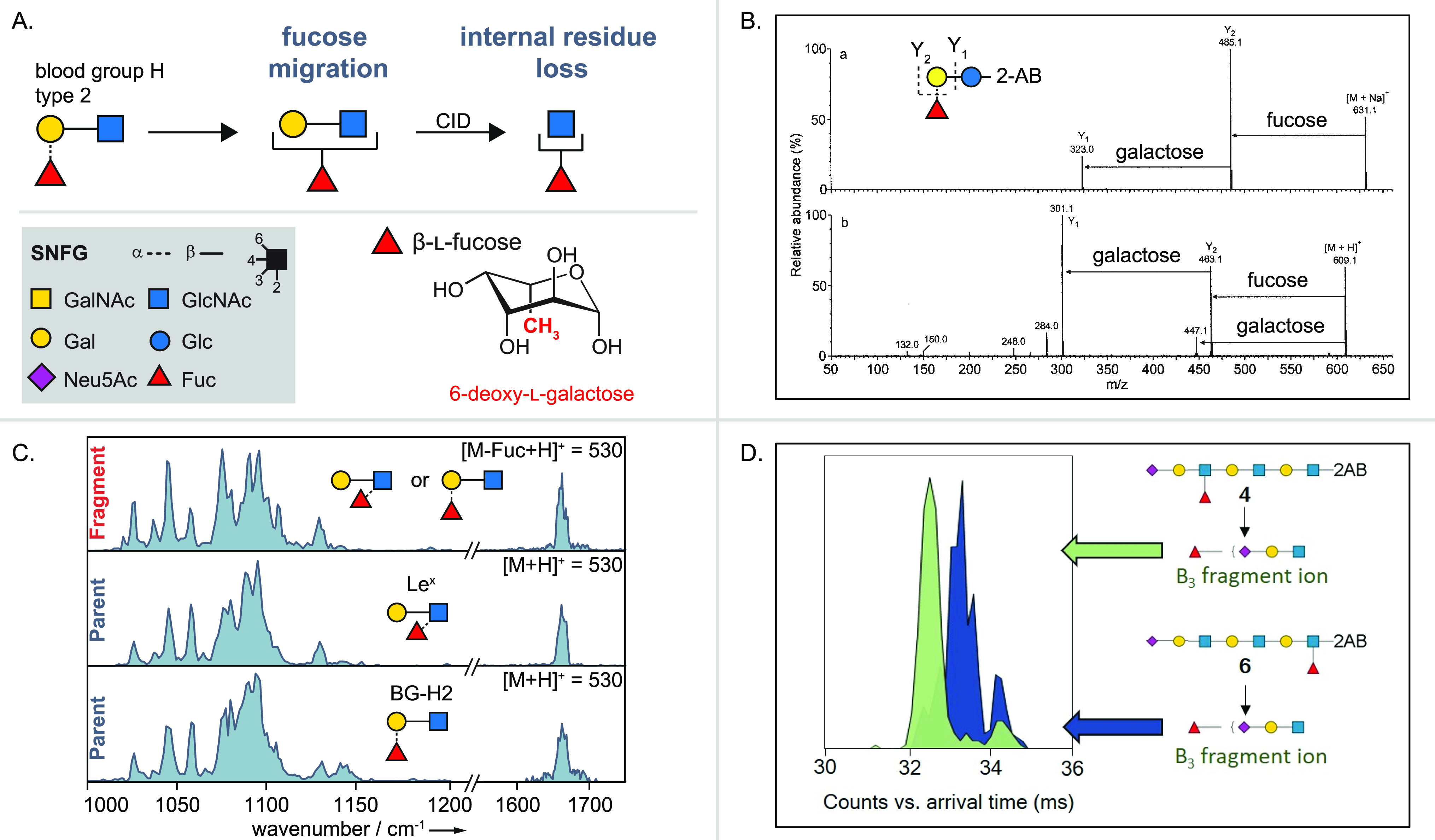
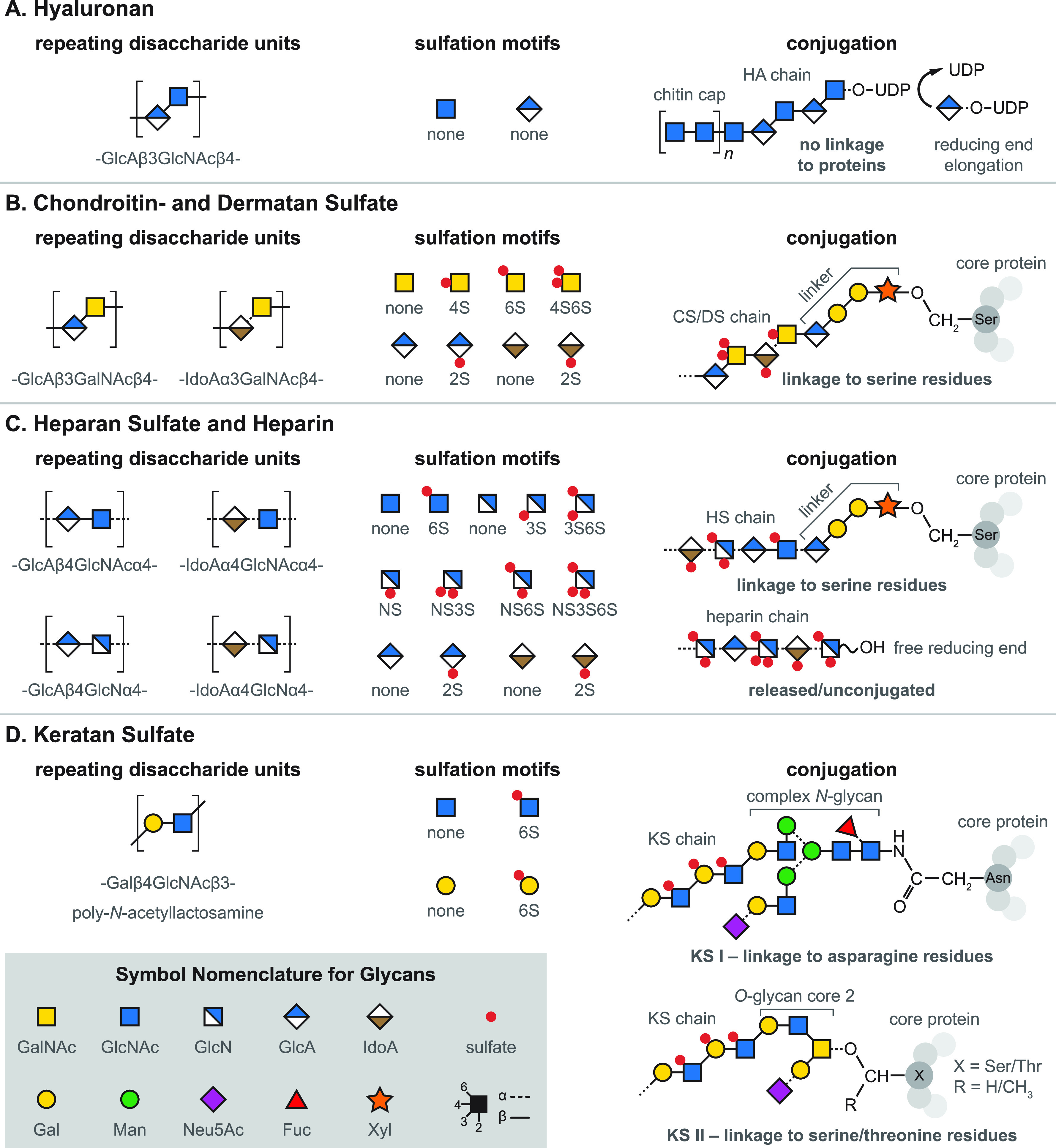
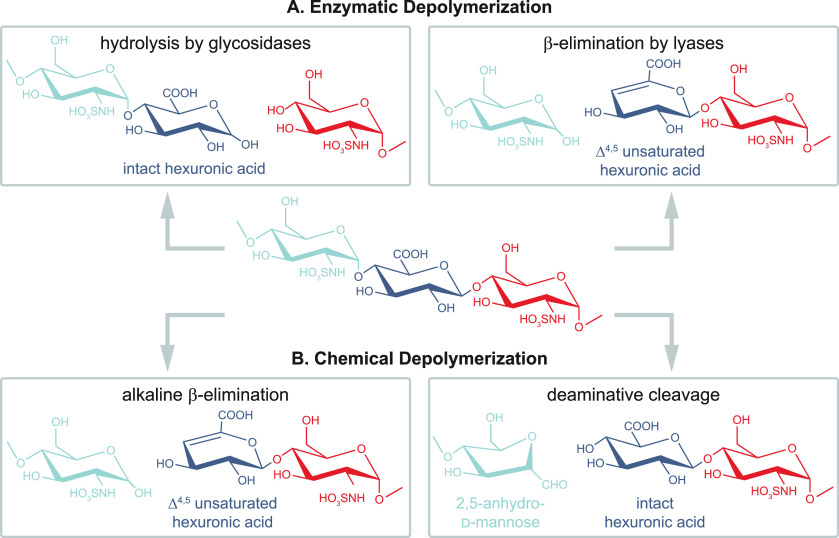
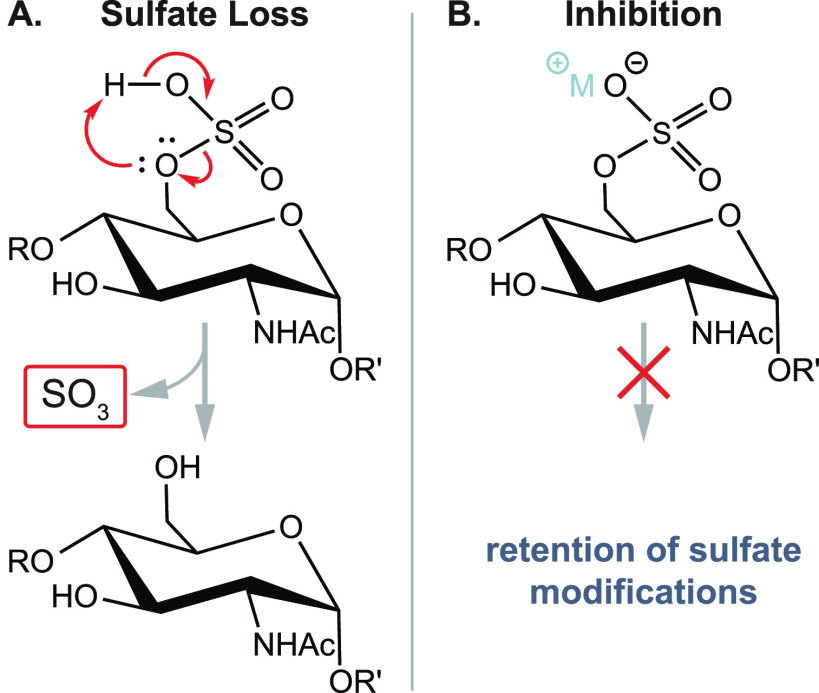
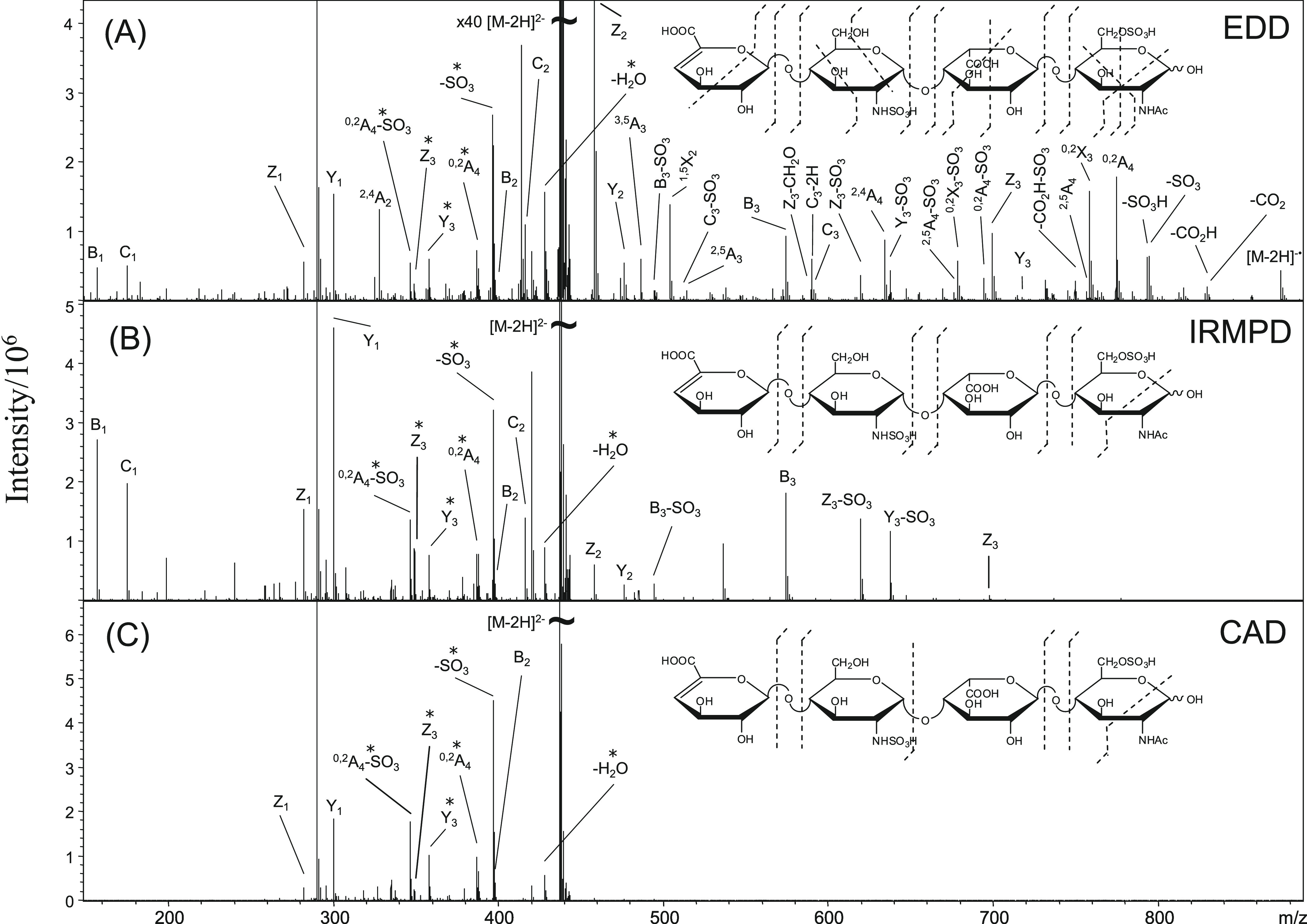
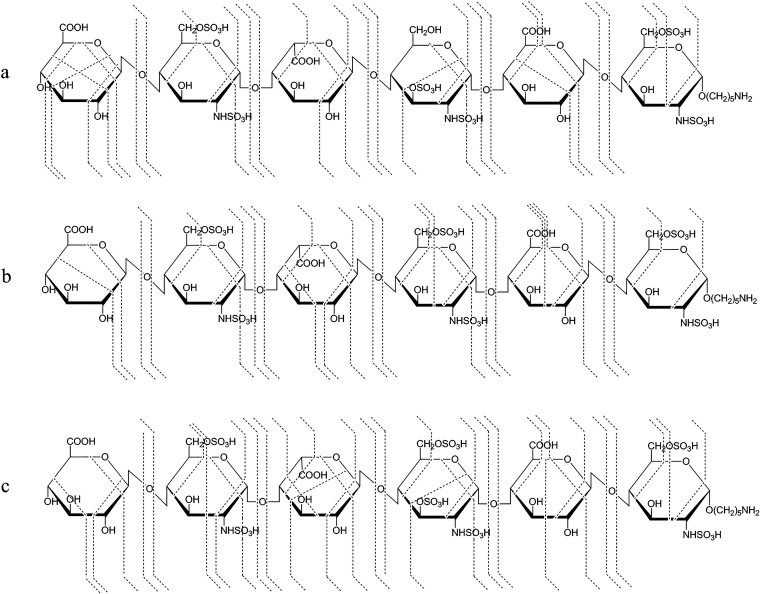
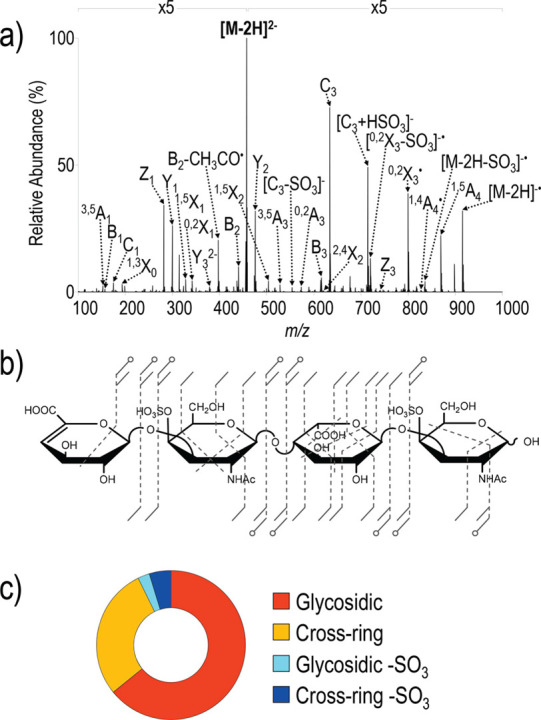
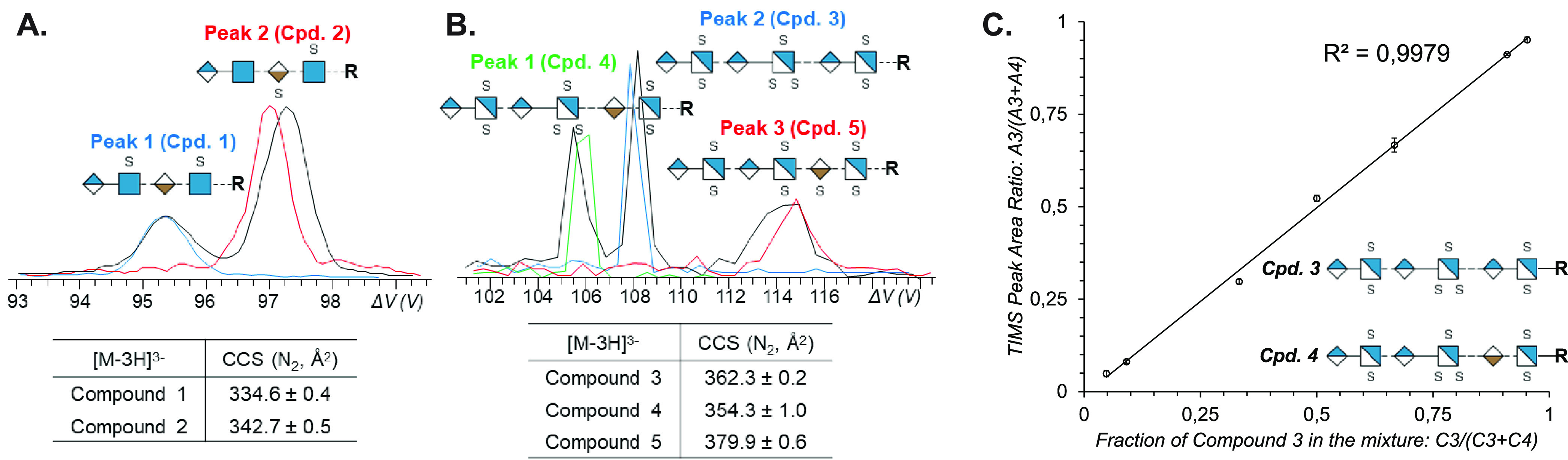
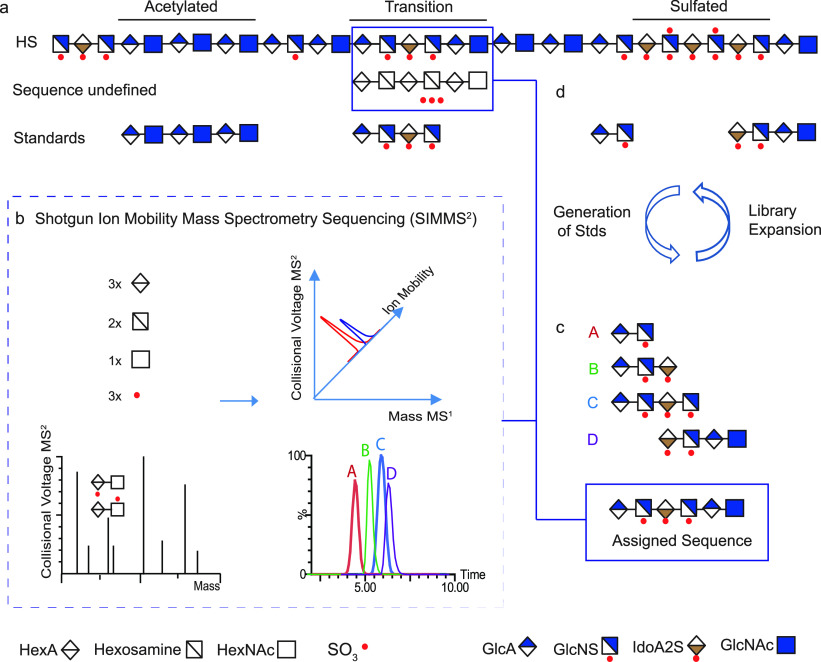
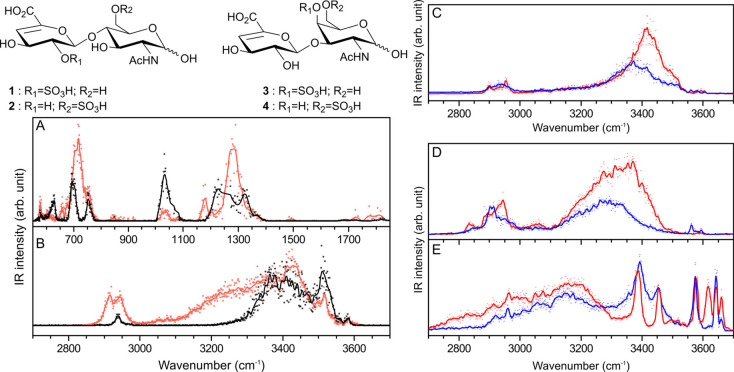
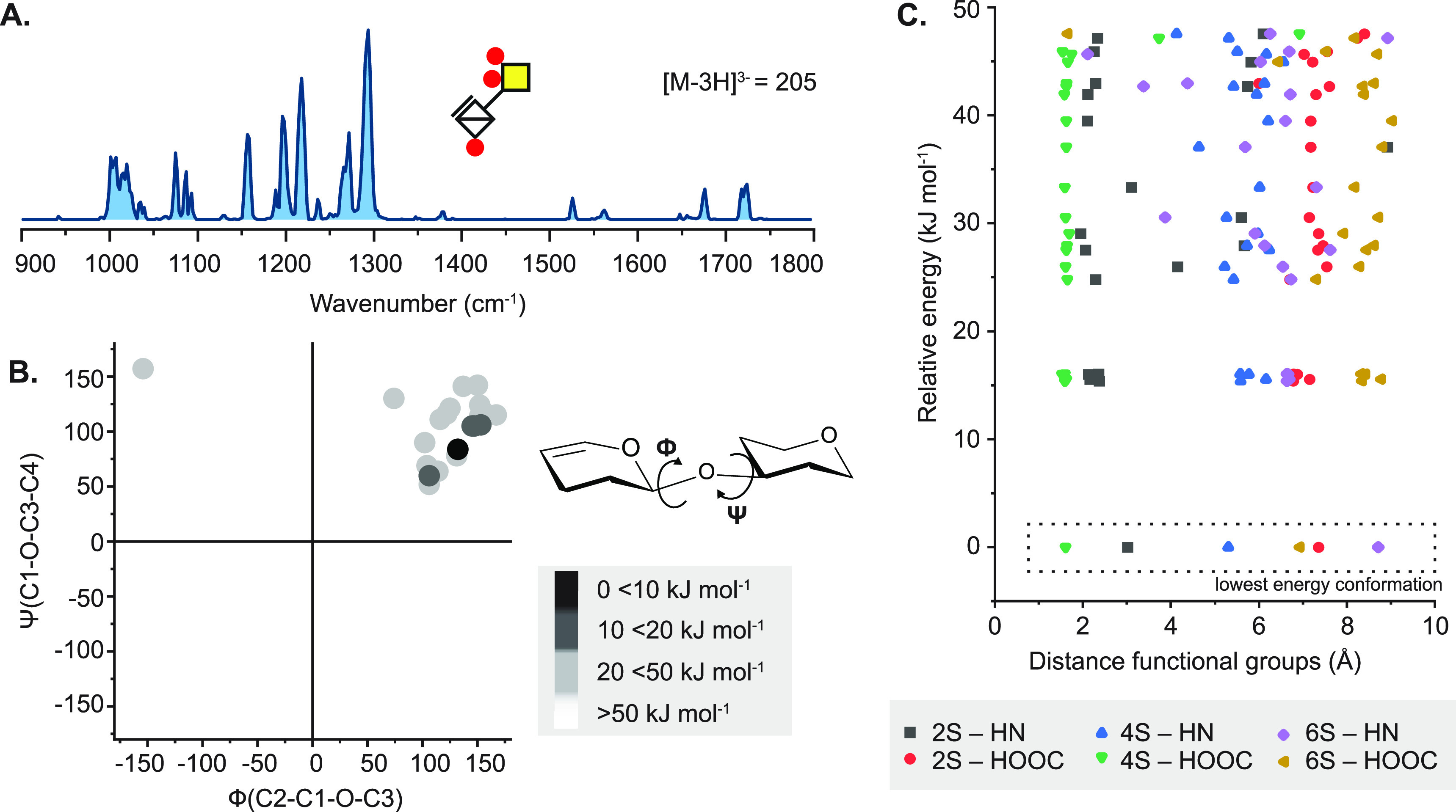
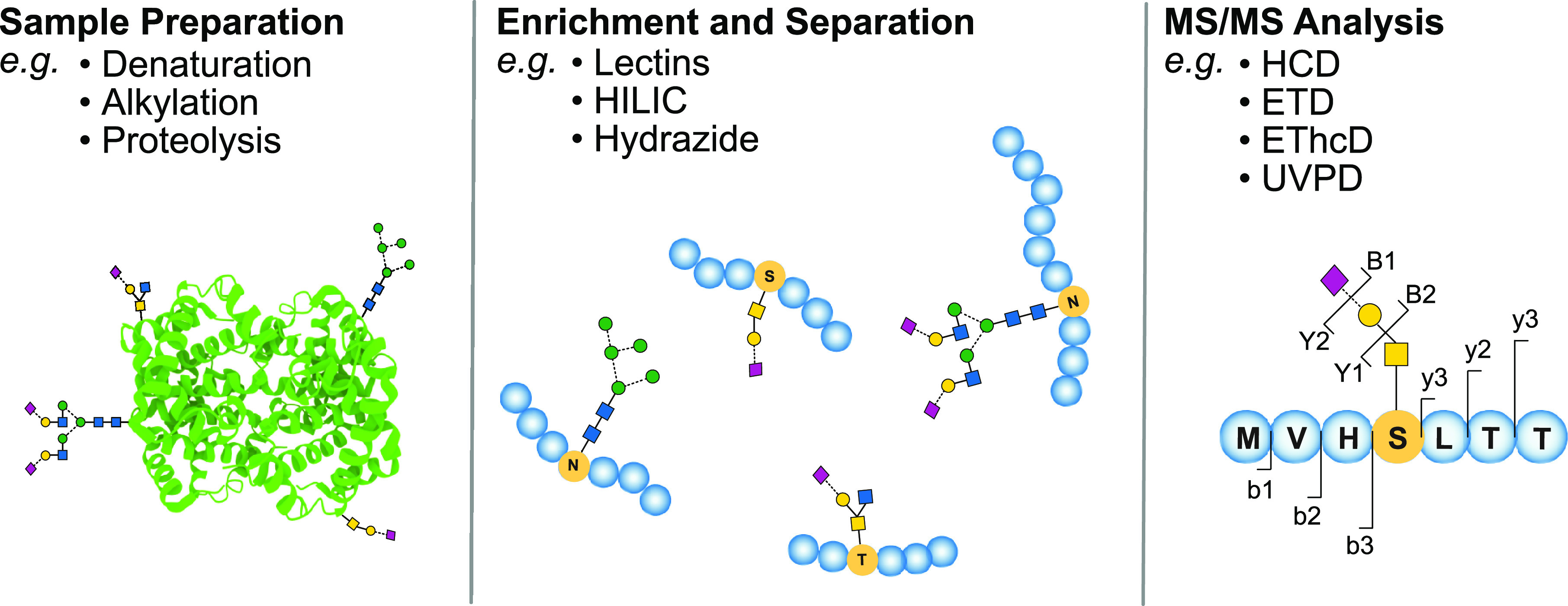
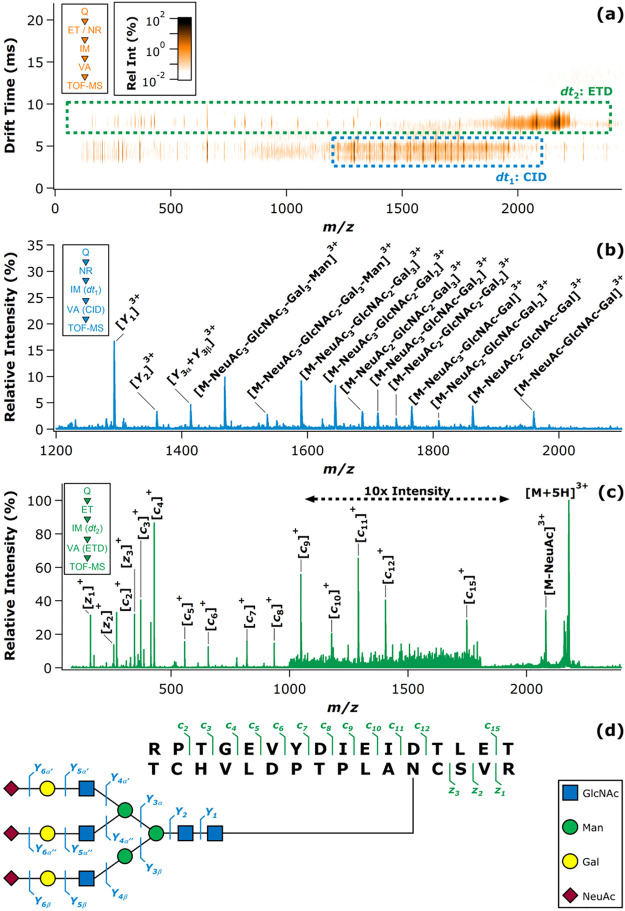
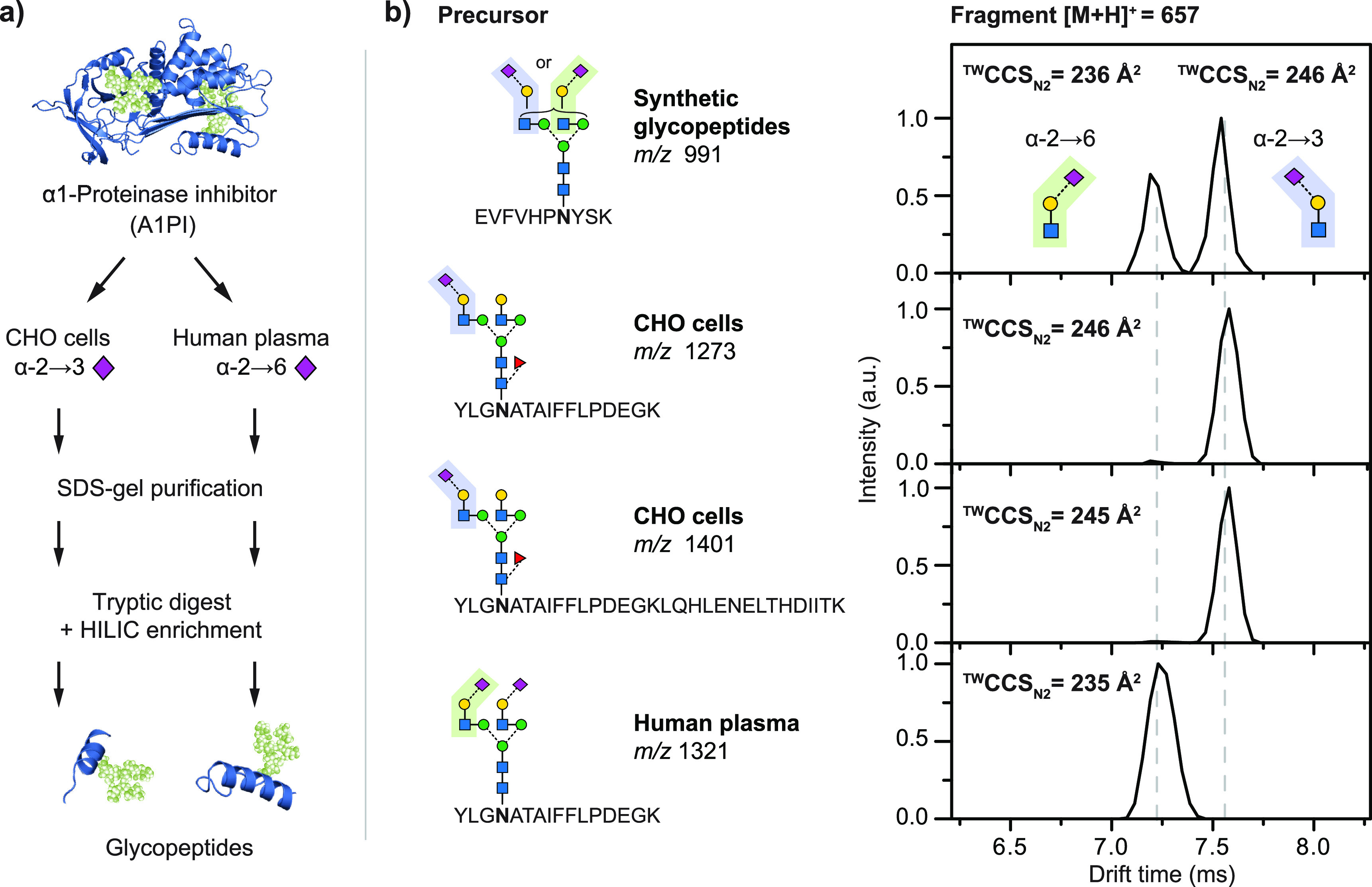
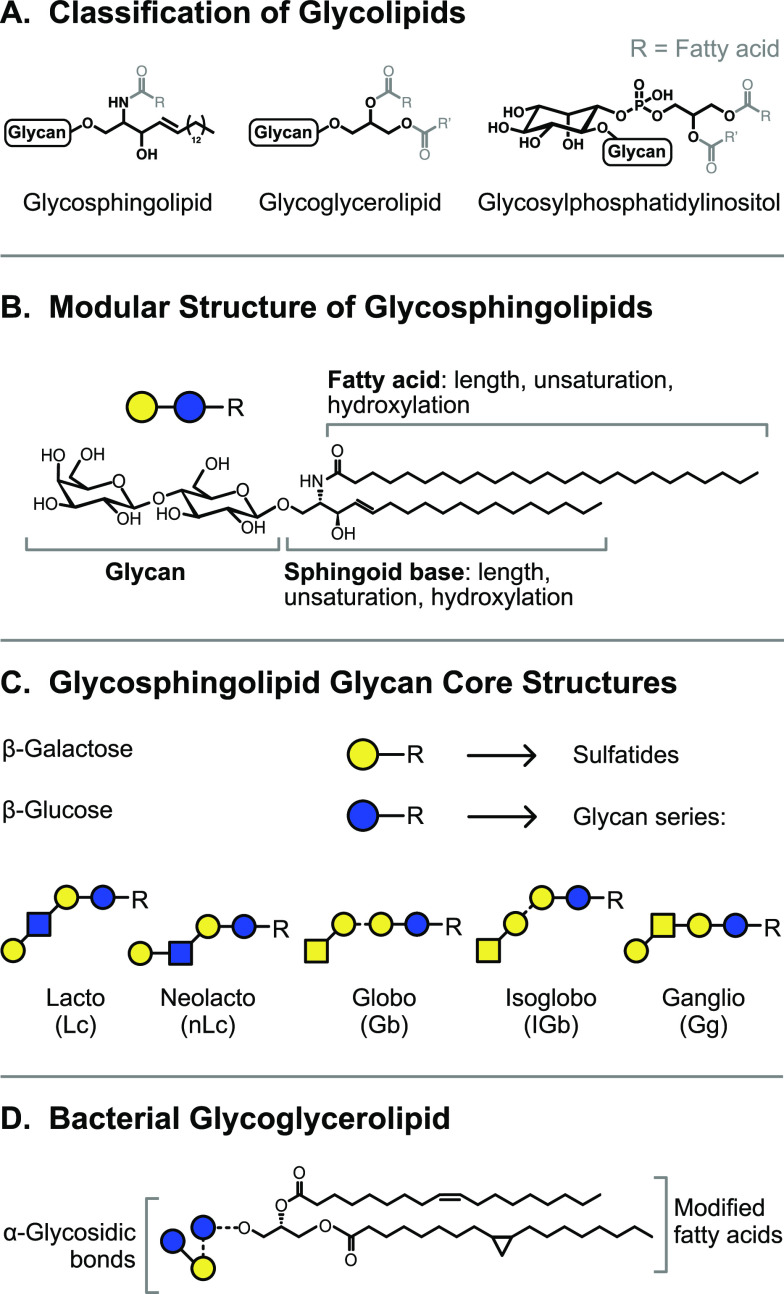
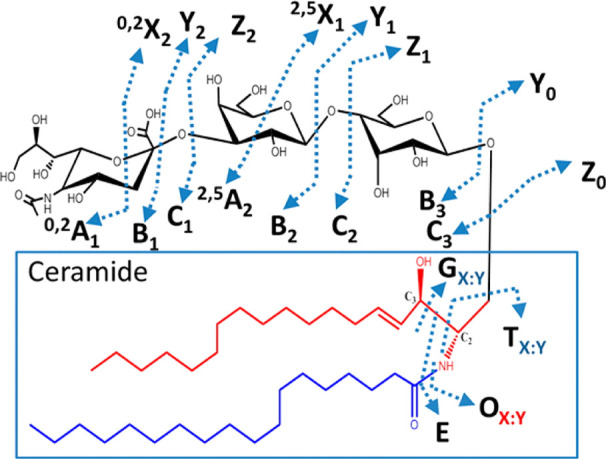
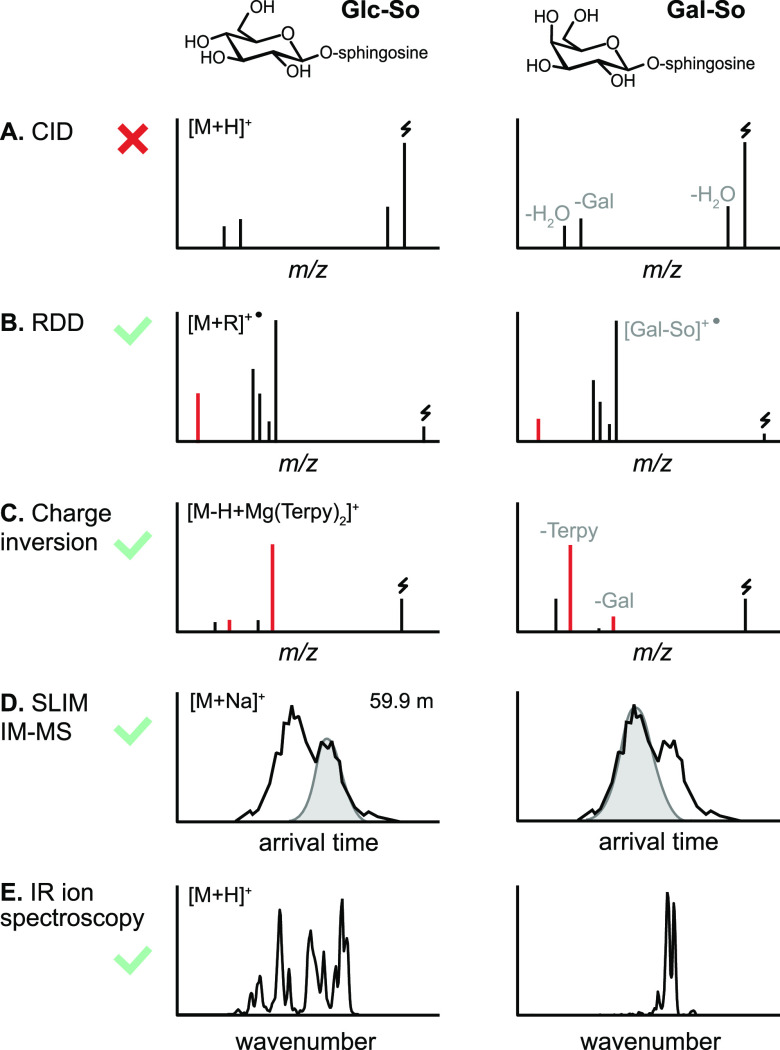
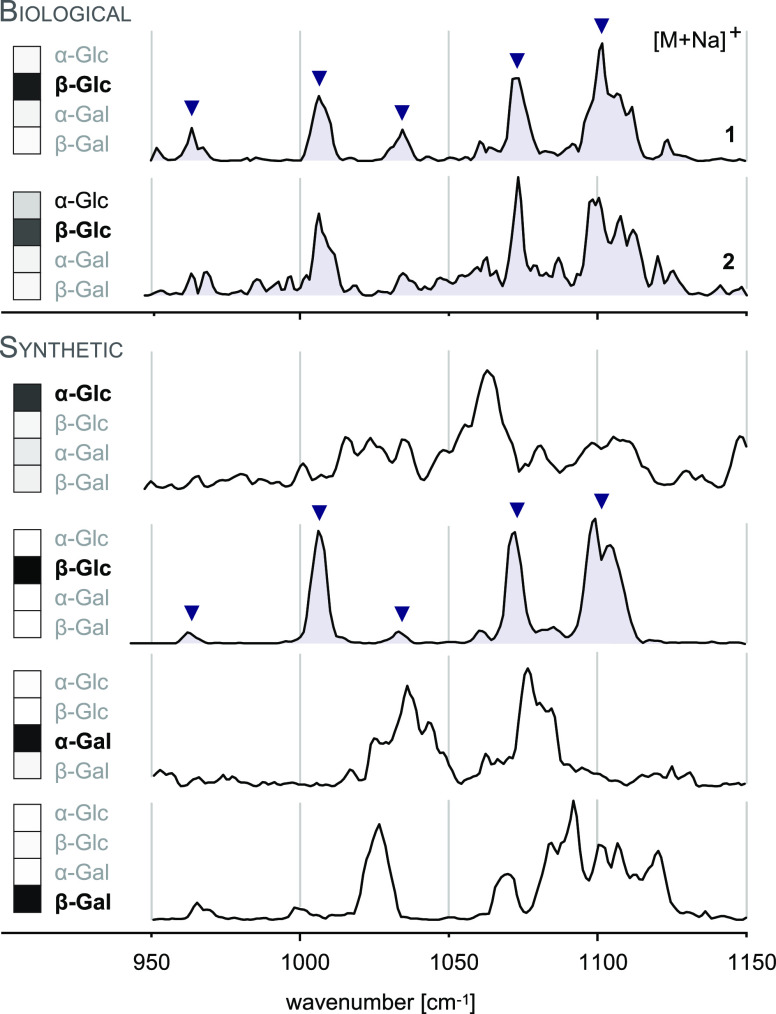
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
