

Blood transcriptomics reveal the evolution and resolution of the immune response in tuberculosis
- PMID: 34491266
- PMCID: PMC8493863
- DOI: 10.1084/jem.20210915
Blood transcriptomics reveal the evolution and resolution of the immune response in tuberculosis
Abstract
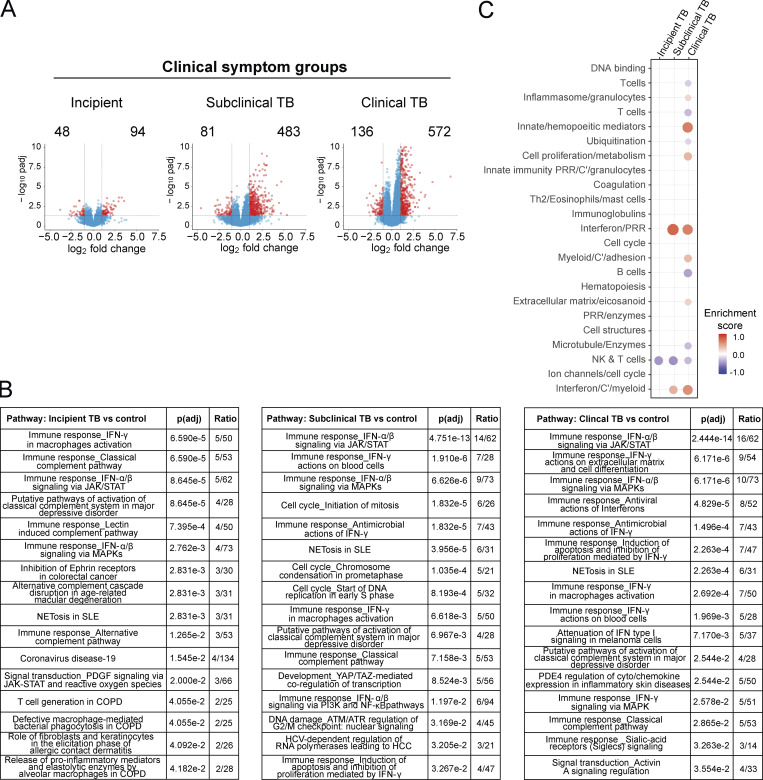
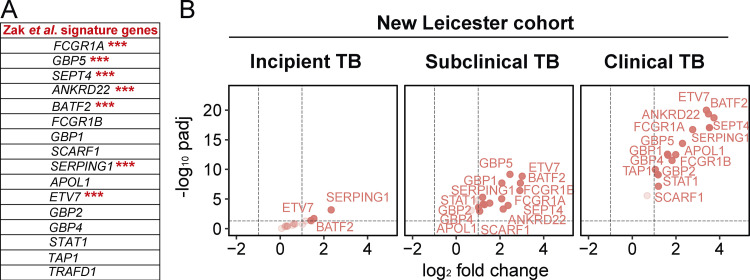
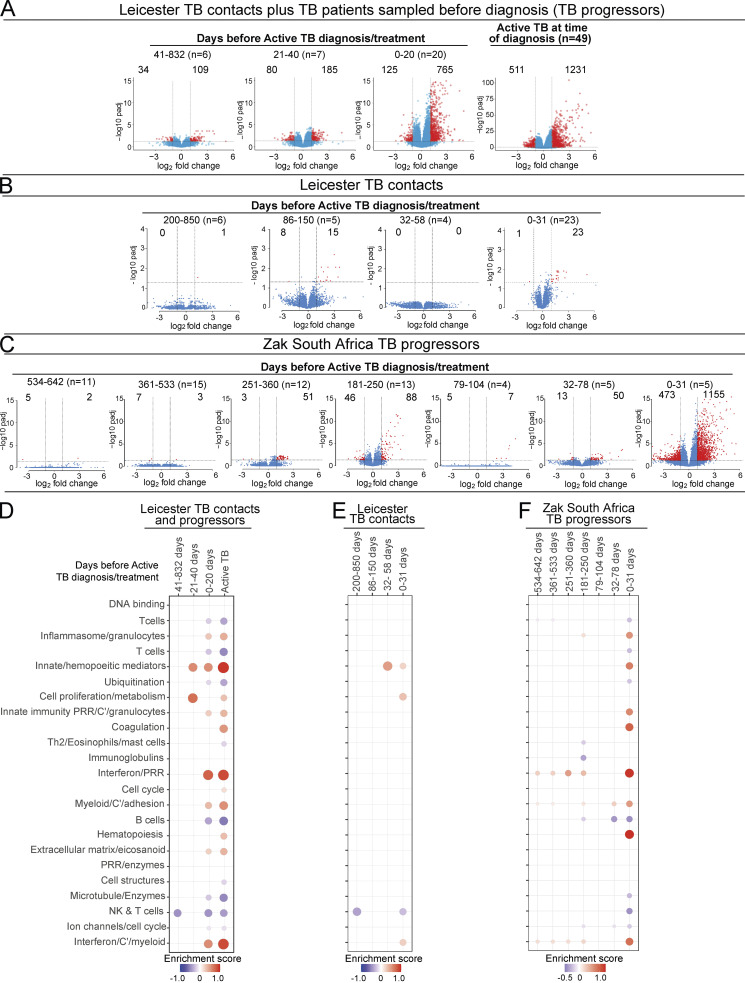
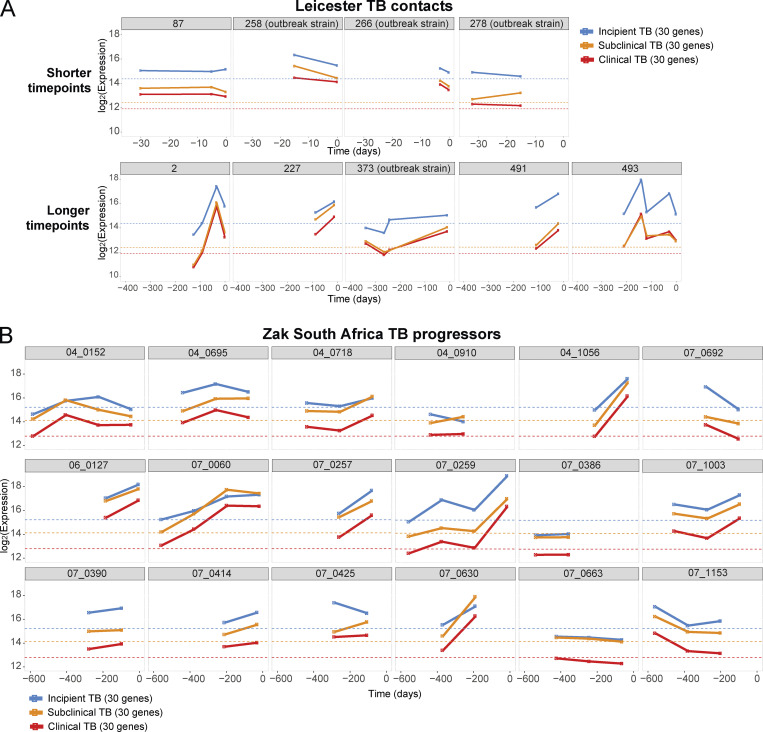
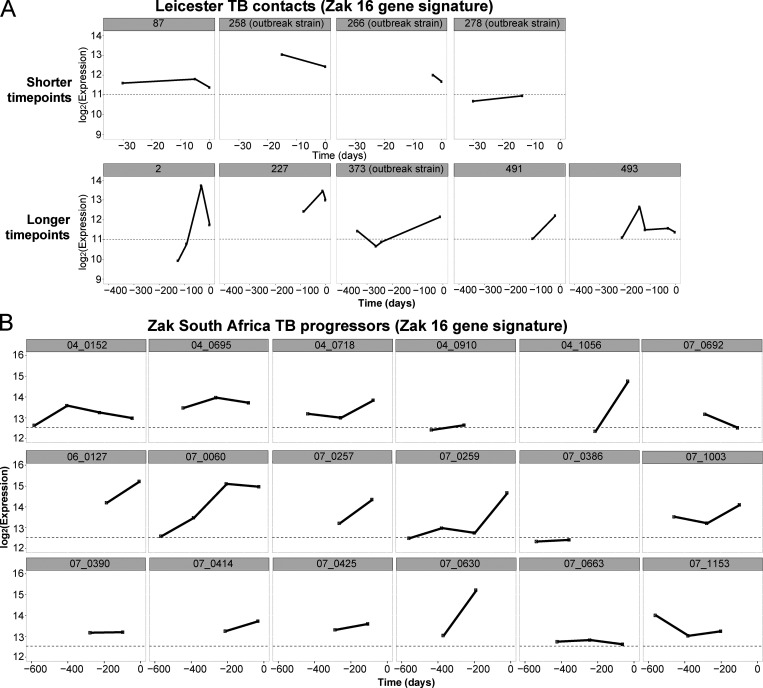
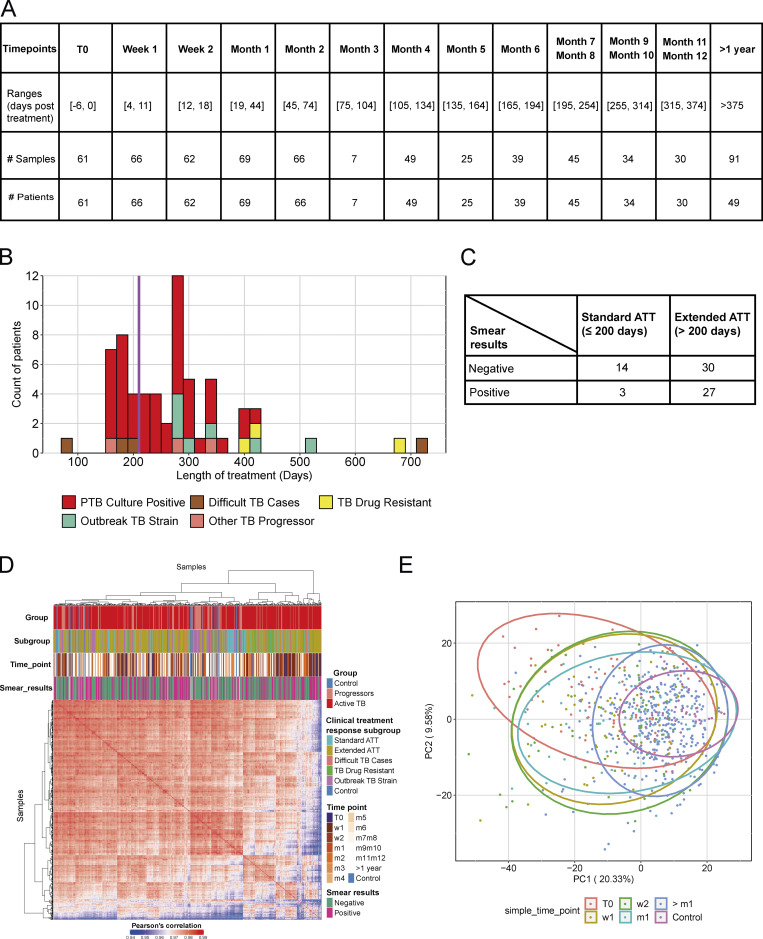
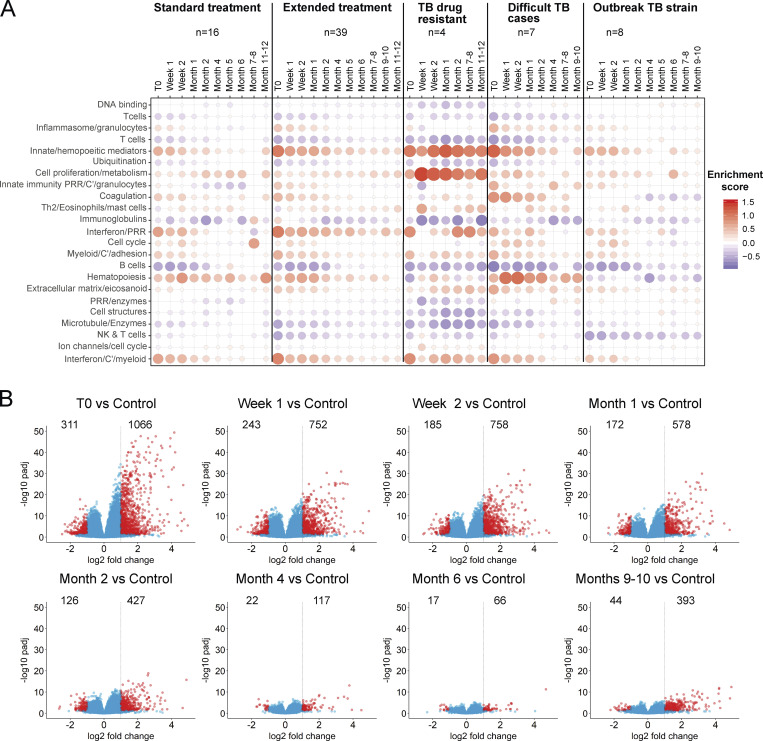
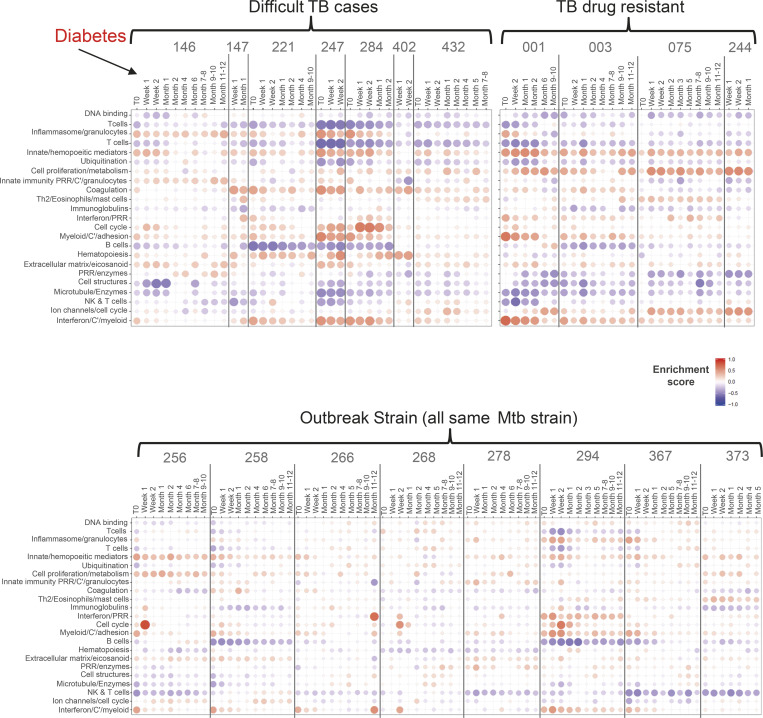
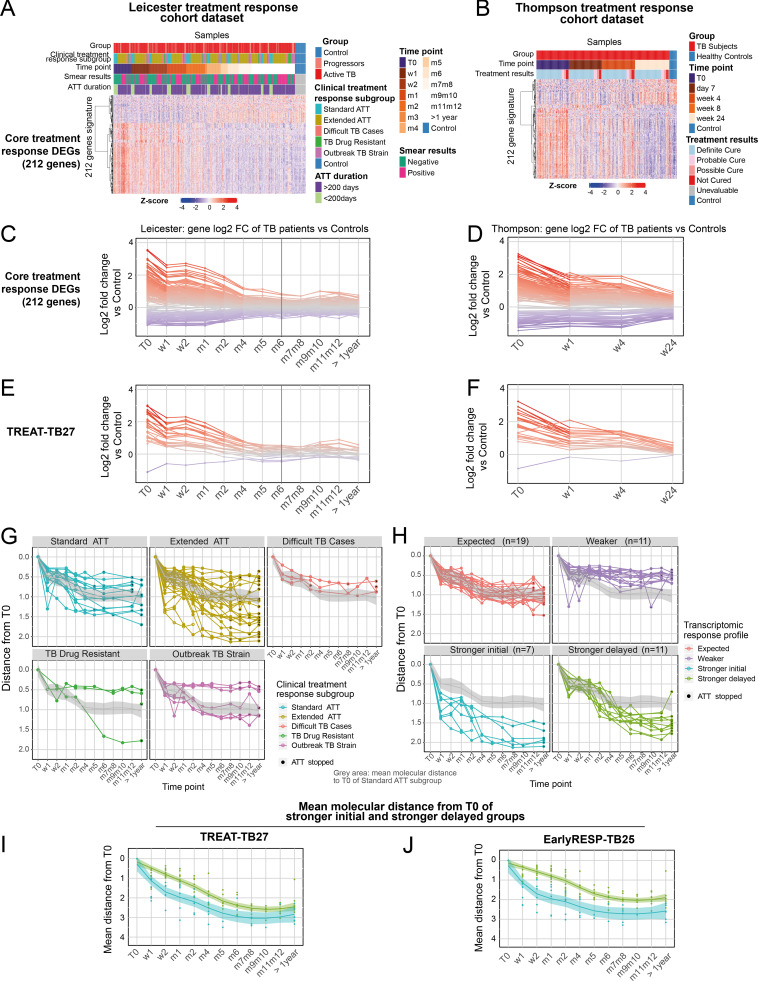
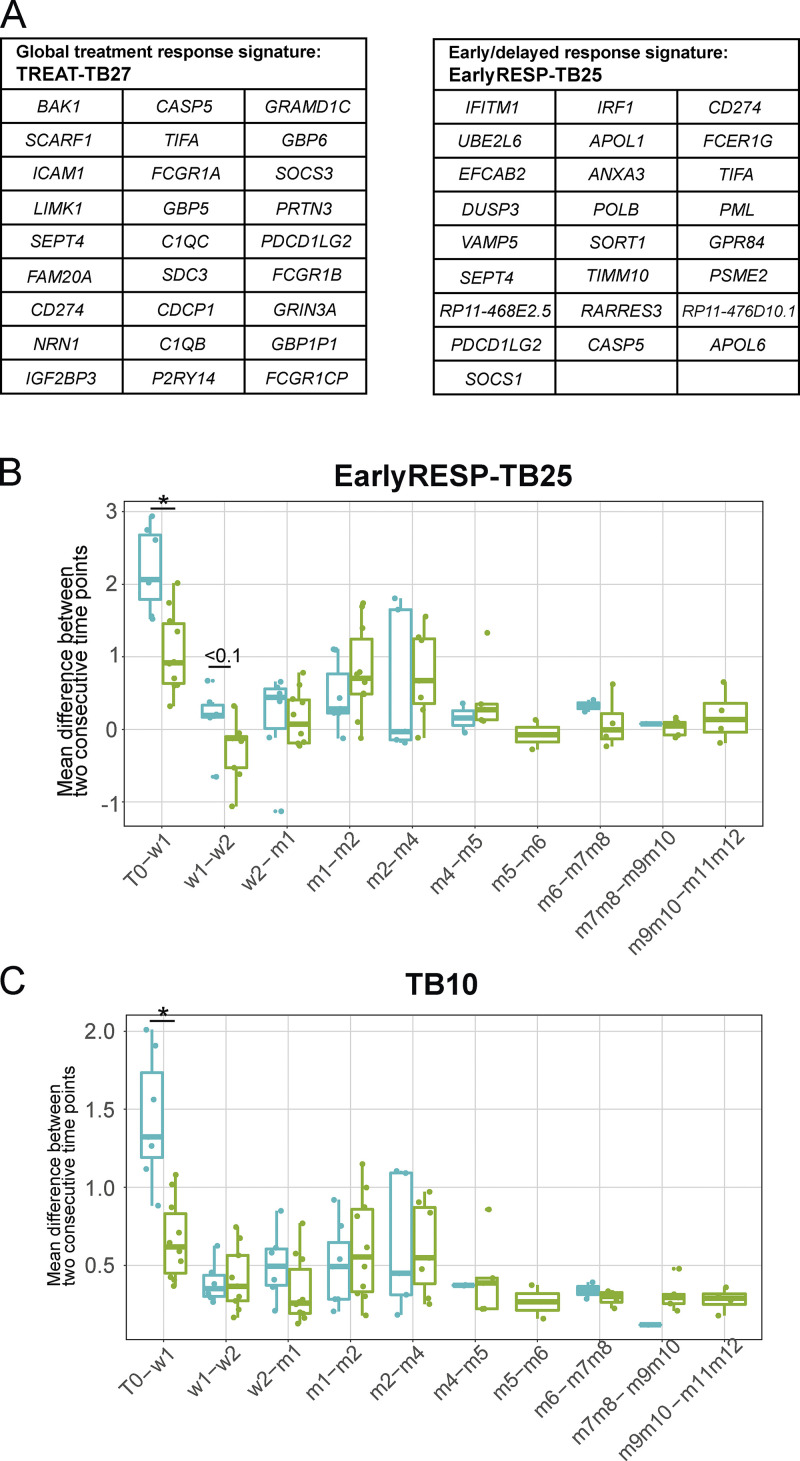
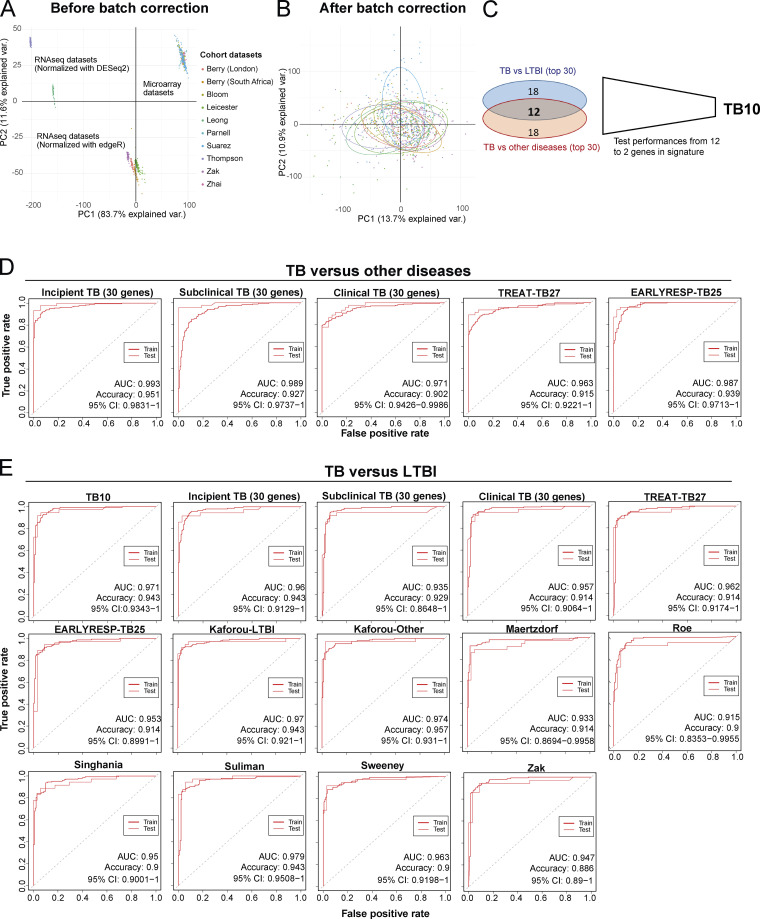
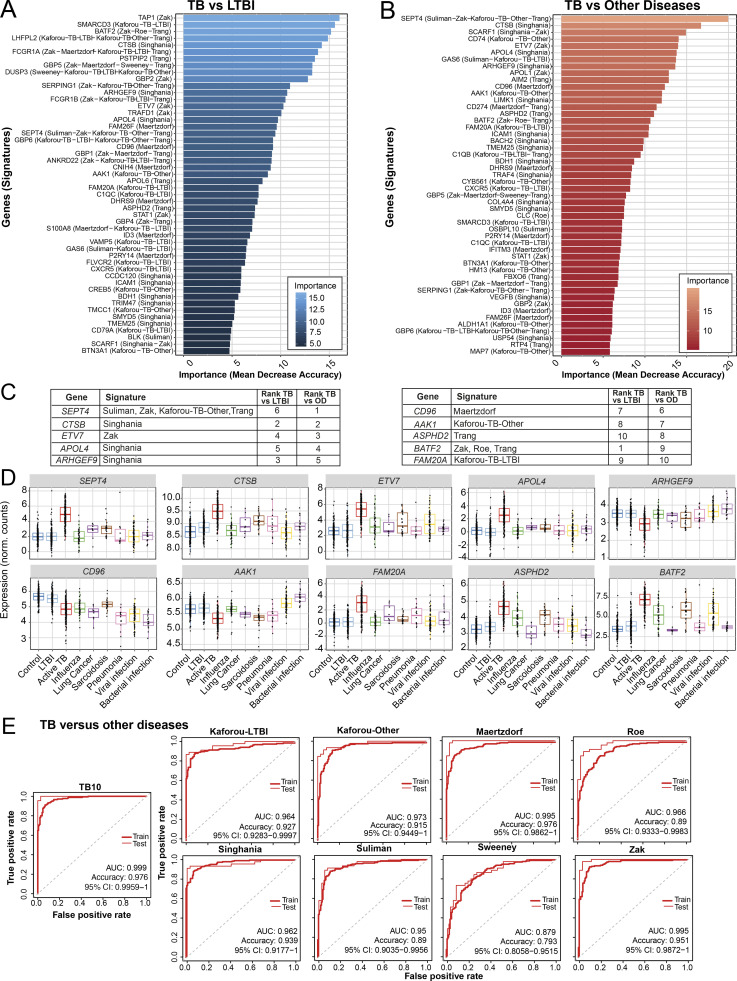
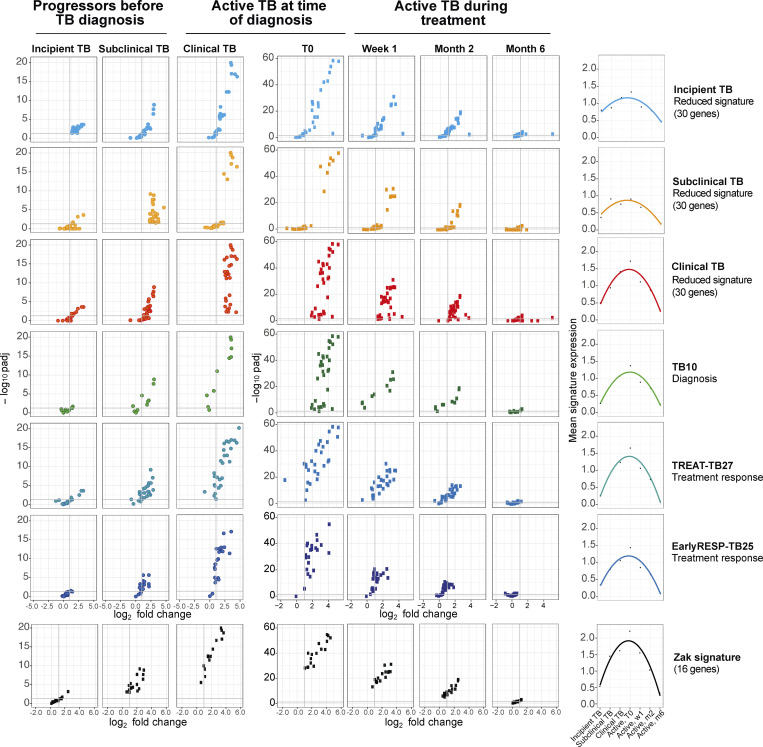
Blood transcriptomics have revealed major characteristics of the immune response in active TB, but the signature early after infection is unknown. In a unique clinically and temporally well-defined cohort of household contacts of active TB patients that progressed to TB, we define minimal changes in gene expression in incipient TB increasing in subclinical and clinical TB. While increasing with time, changes in gene expression were highest at 30 d before diagnosis, with heterogeneity in the response in household TB contacts and in a published cohort of TB progressors as they progressed to TB, at a bulk cohort level and in individual progressors. Blood signatures from patients before and during anti-TB treatment robustly monitored the treatment response distinguishing early and late responders. Blood transcriptomics thus reveal the evolution and resolution of the immune response in TB, which may help in clinical management of the disease.
© 2021 Tabone et al.
Conflict of interest statement
Disclosures: W.J. Branchett reported their postdoctoral position is funded by a Wellcome Trust Investigator Award to Professor Anne O'Garra. M. Rodrigue reported they are a bioMérieux employee. No other disclosures were reported.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
