

C-H Bond Cleavage by Bioinspired Nonheme Metal Complexes
- PMID: 34491738
- PMCID: PMC8590853
- DOI: 10.1021/acs.inorgchem.1c01754
C-H Bond Cleavage by Bioinspired Nonheme Metal Complexes
Abstract
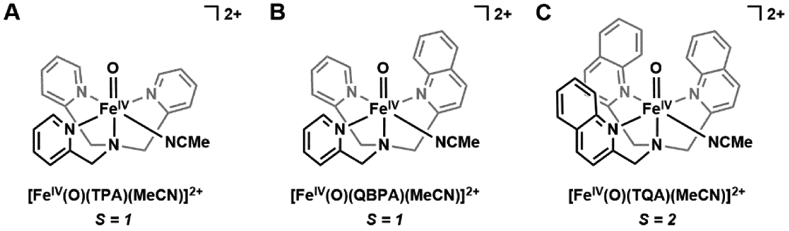
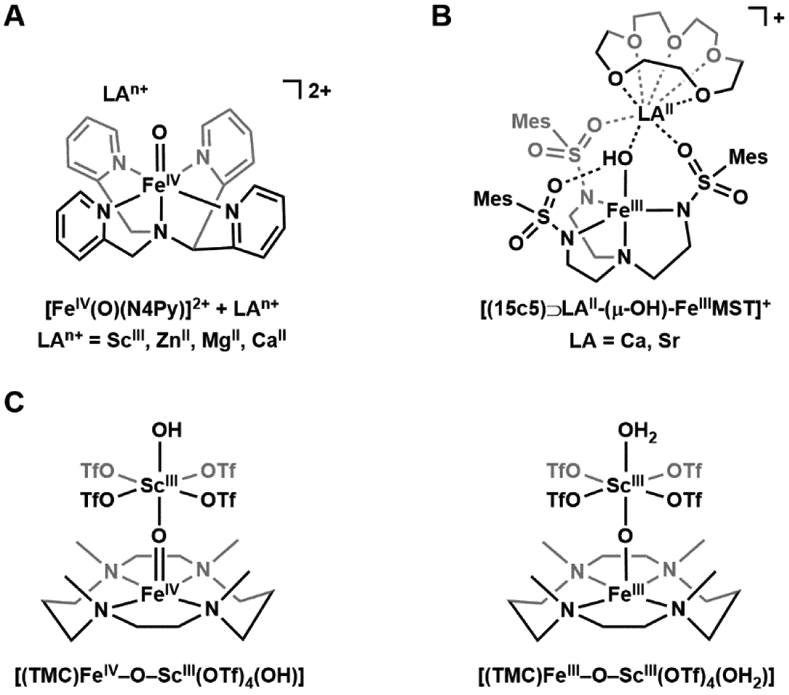
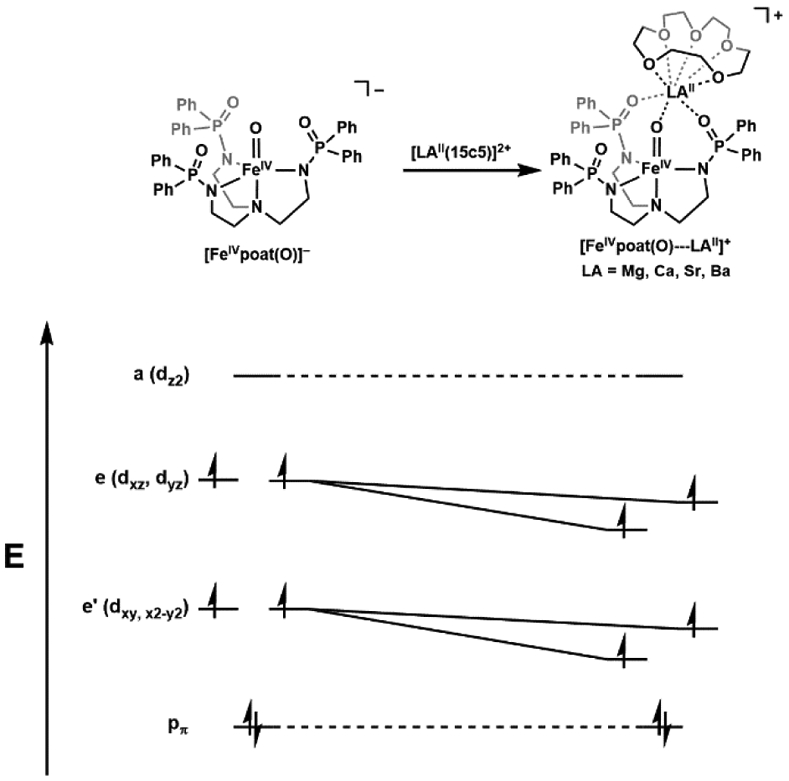
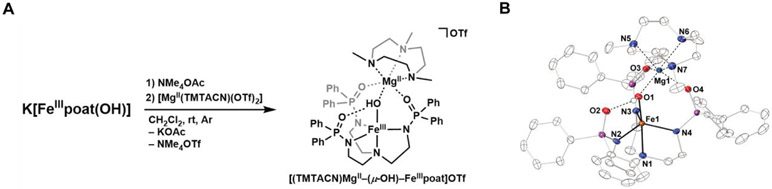
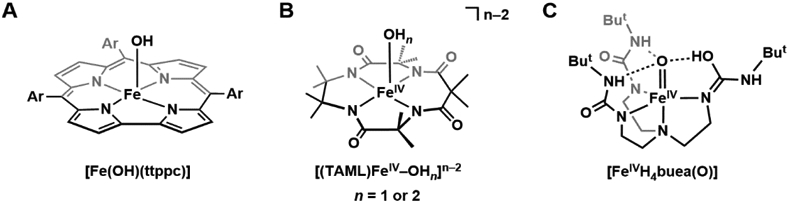
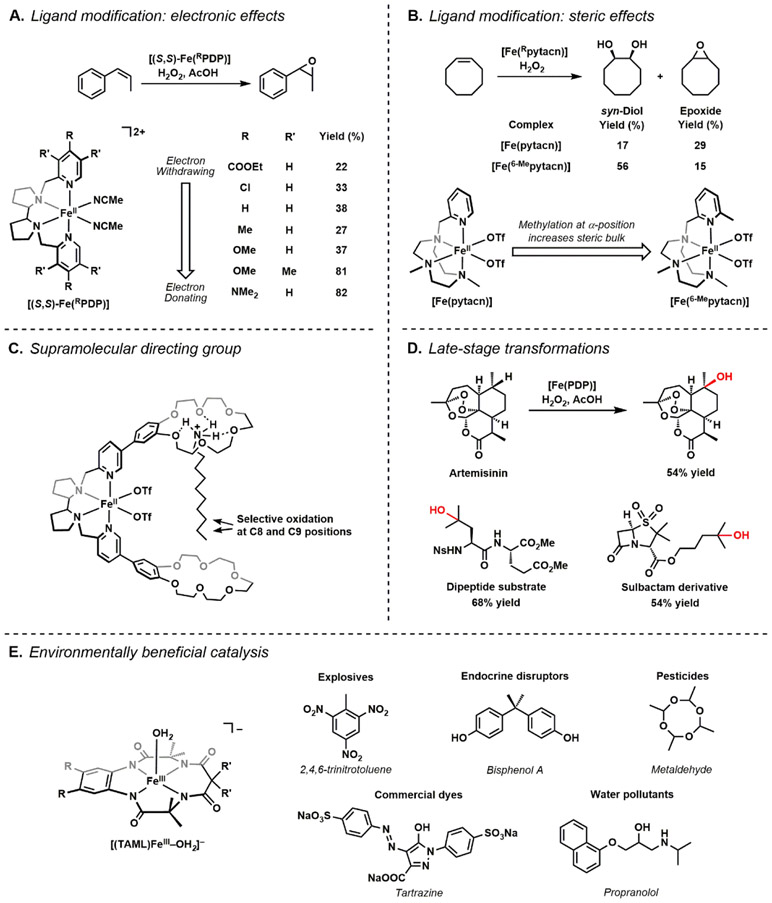
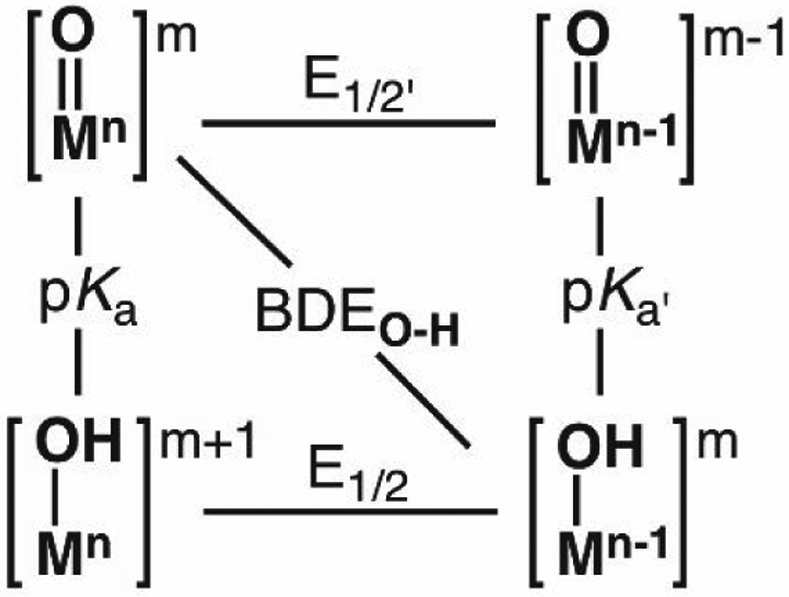
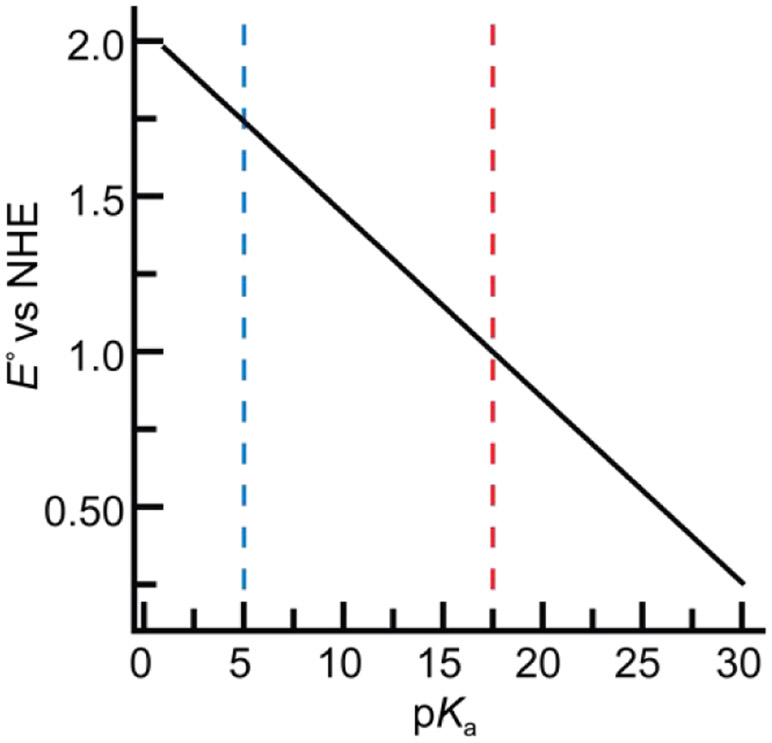
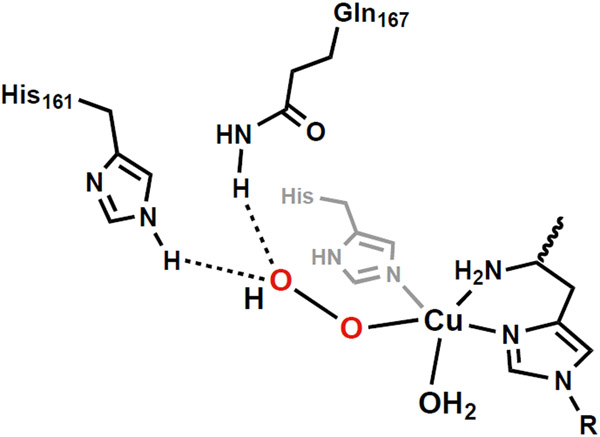
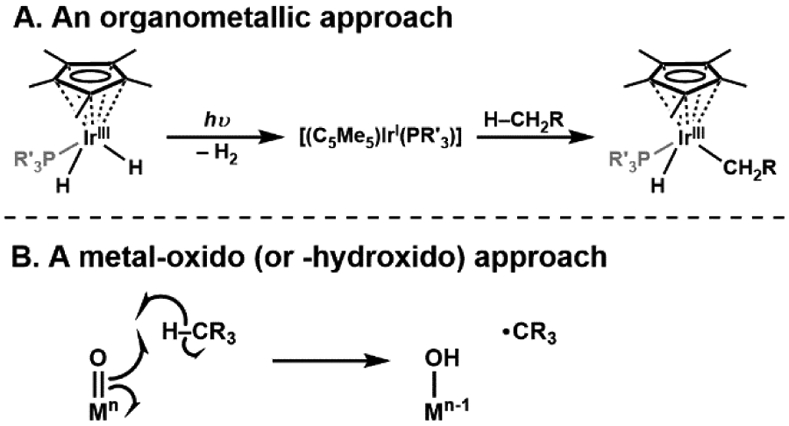
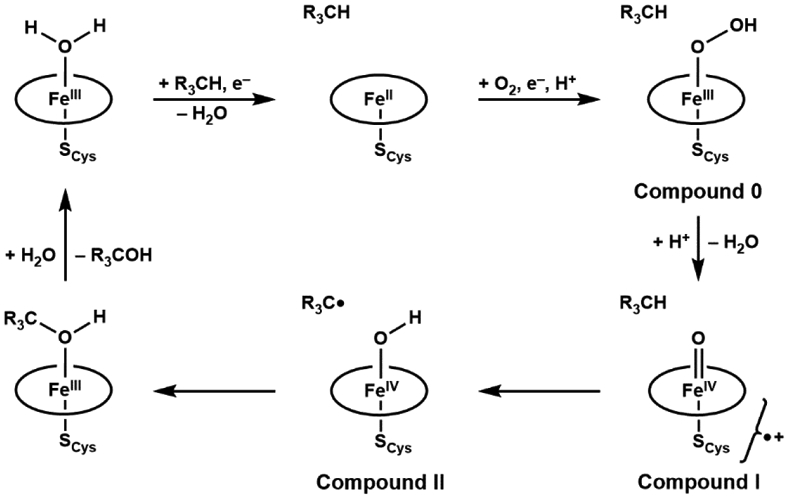
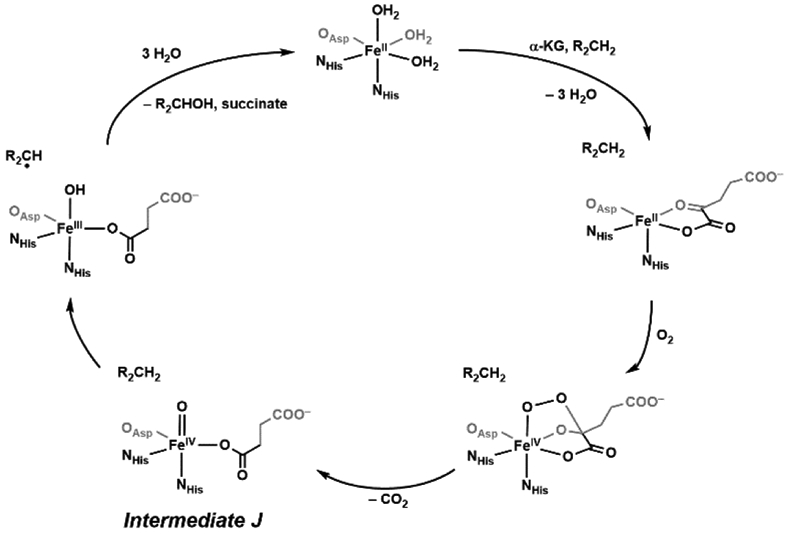
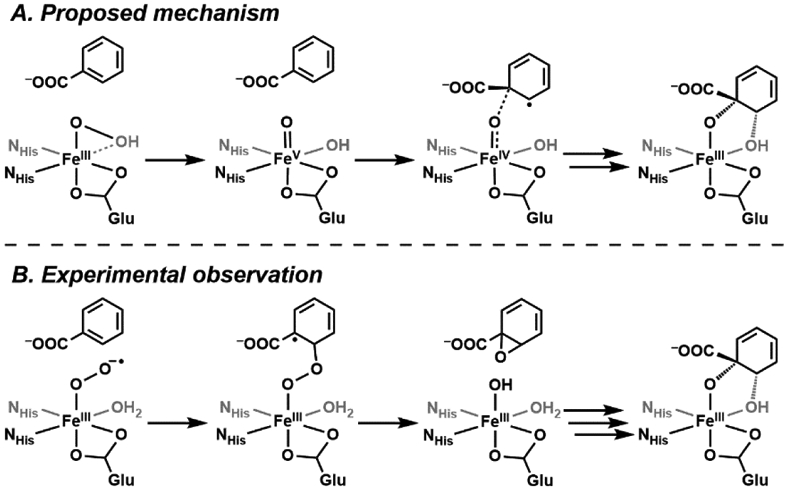
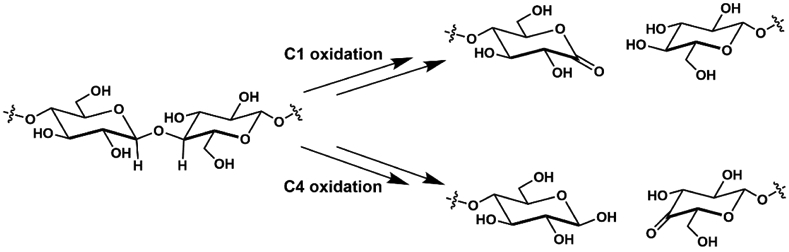
The functionalization of C-H bonds is one of the most challenging transformations in synthetic chemistry. In biology, these processes are well-known and are achieved with a variety of metalloenzymes, many of which contain a single metal center within their active sites. The most well studied are those with Fe centers, and the emerging experimental data show that high-valent iron oxido species are the intermediates responsible for cleaving the C-H bond. This Forum Article describes the state of this field with an emphasis on nonheme Fe enzymes and current experimental results that provide insights into the properties that make these species capable of C-H bond cleavage. These parameters are also briefly considered in regard to manganese oxido complexes and Cu-containing metalloenzymes. Synthetic iron oxido complexes are discussed to highlight their utility as spectroscopic and mechanistic probes and reagents for C-H bond functionalization. Avenues for future research are also examined.
Figures
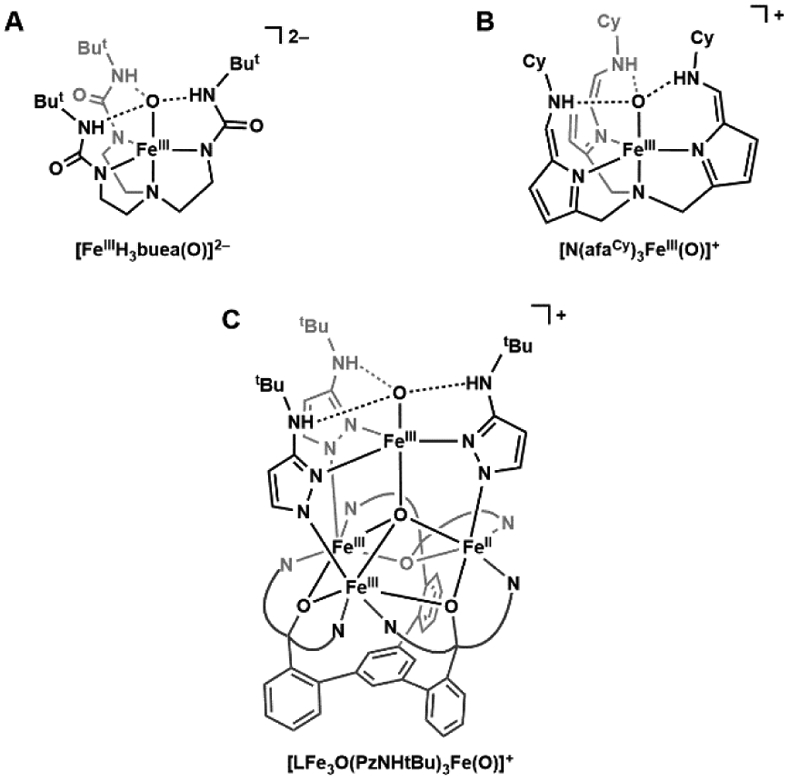
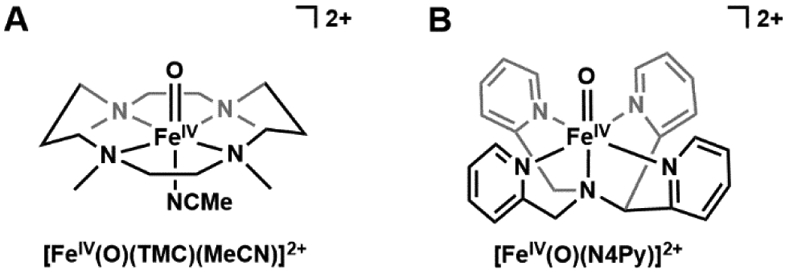
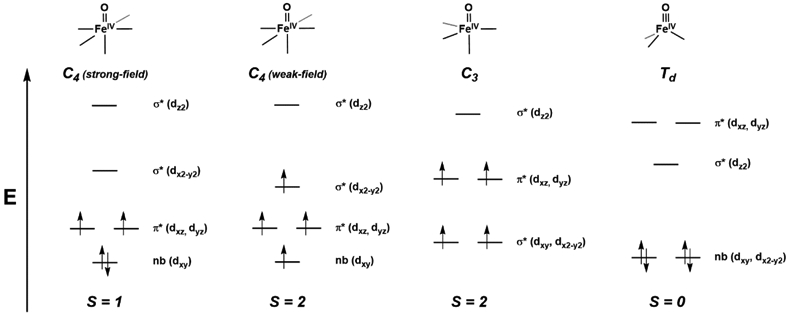
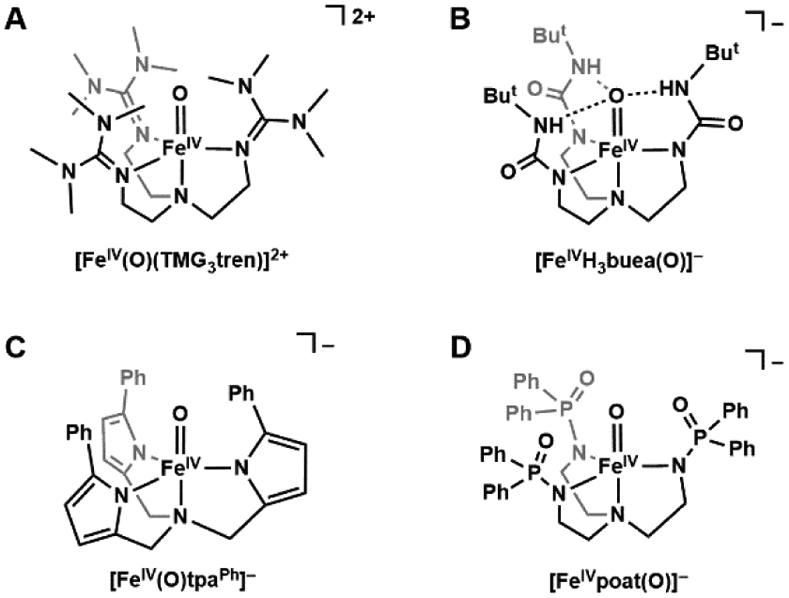
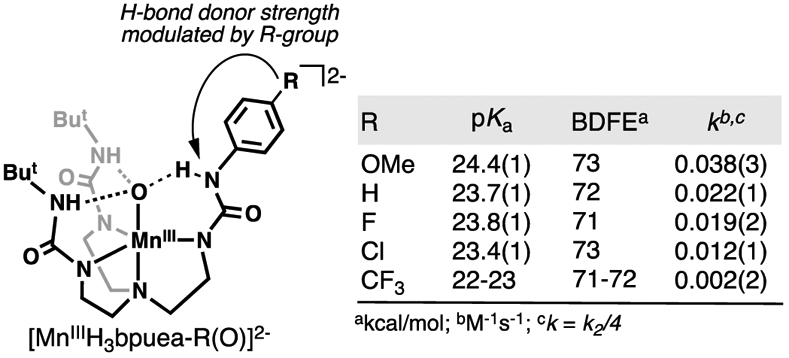
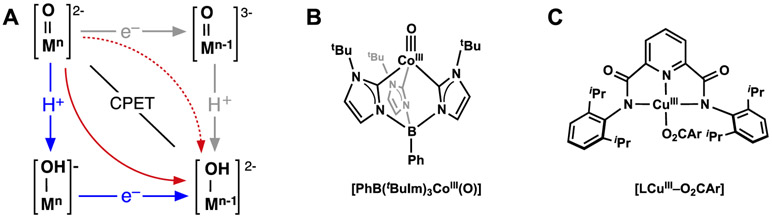
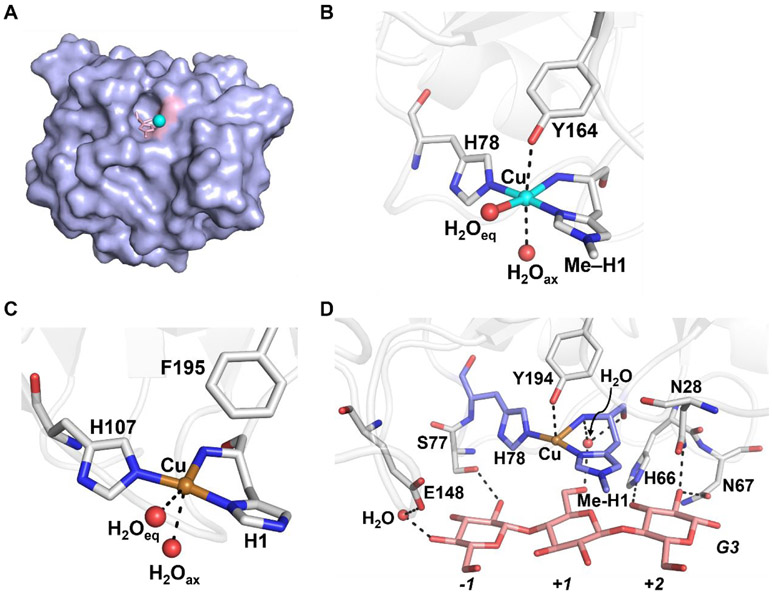
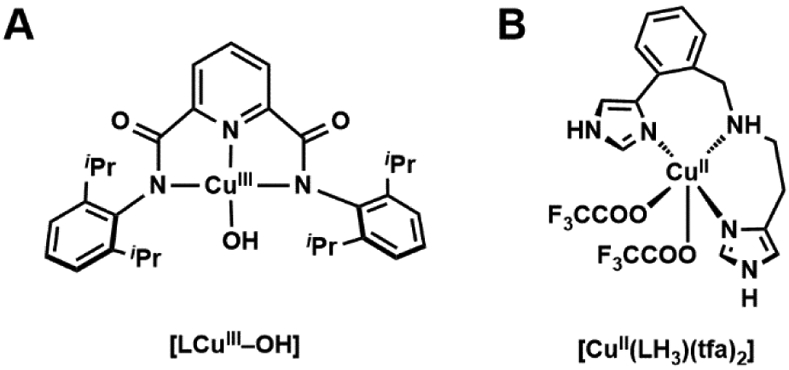
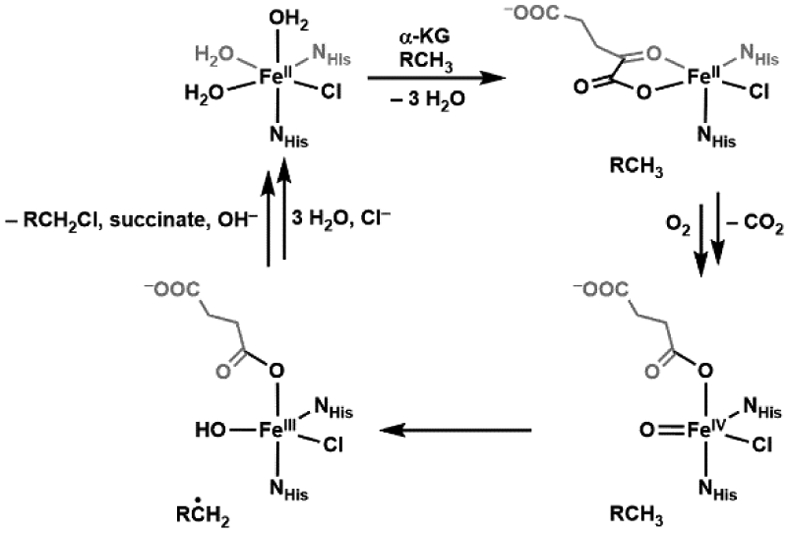
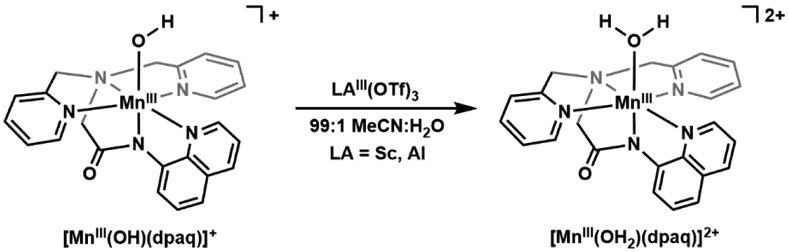
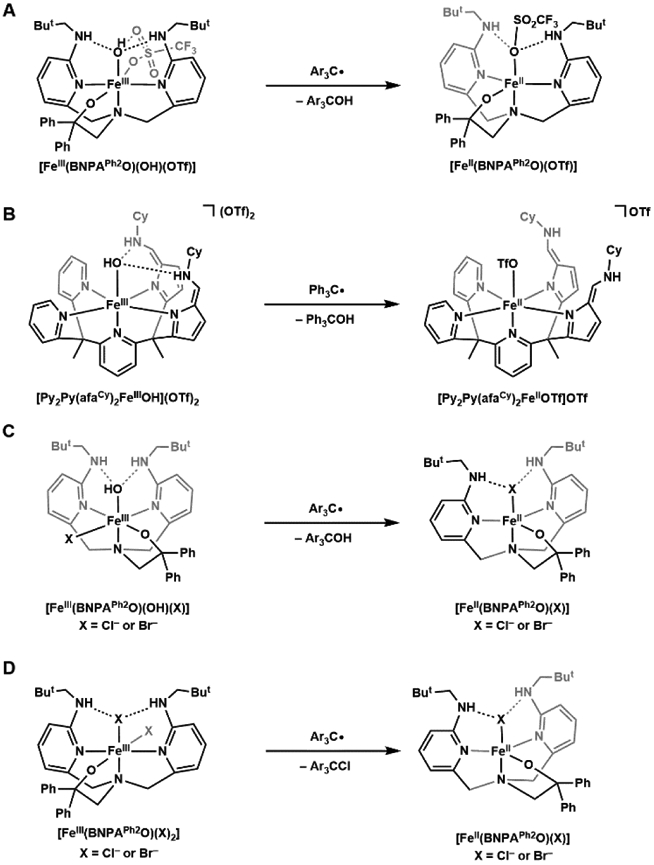
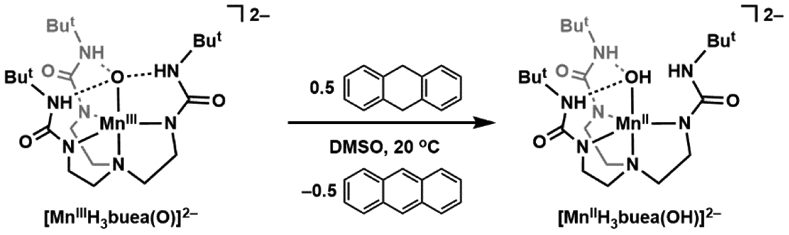
References
-
- Que L; Tolman WB Biologically Inspired Oxidation Catalysis. Nature 2008, 455, 333–340. - PubMed
-
- Arndtsen BA; Bergman RG; Mobley TA; Peterson TH Selective Intermolecular Carbon–Hydrogen Bond Activation by Synthetic Metal Complexes in Homogeneous Solution. Acc. Chem. Res 1995, 28, 154–162.
-
- Thirunavukkarasu VS; Kozhushkov SI; Ackermann L C─H Nitrogenation and Oxygenation by Ruthenium Catalysis. Chem. Commun 2014, 50, 29–39. - PubMed
