

The antiviral state has shaped the CpG composition of the vertebrate interferome to avoid self-targeting
- PMID: 34491982
- PMCID: PMC8423302
- DOI: 10.1371/journal.pbio.3001352
The antiviral state has shaped the CpG composition of the vertebrate interferome to avoid self-targeting
Abstract
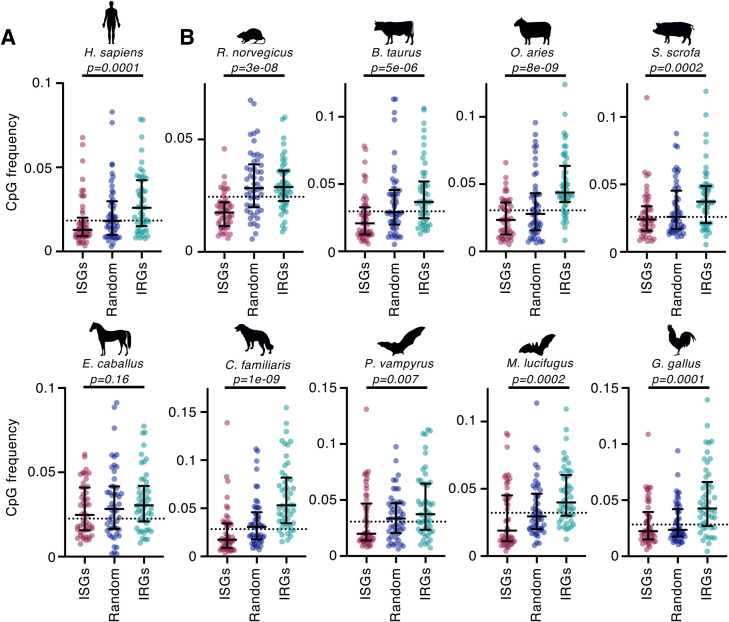
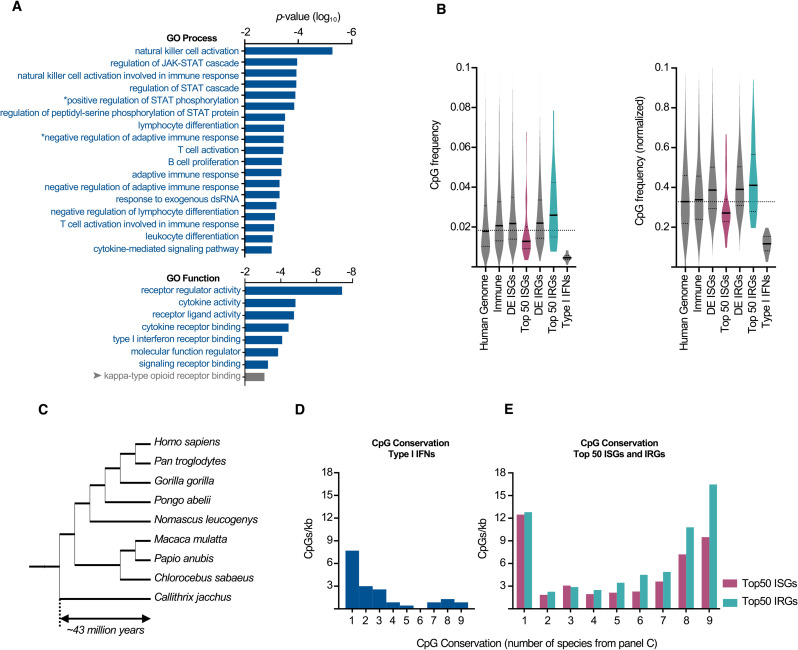
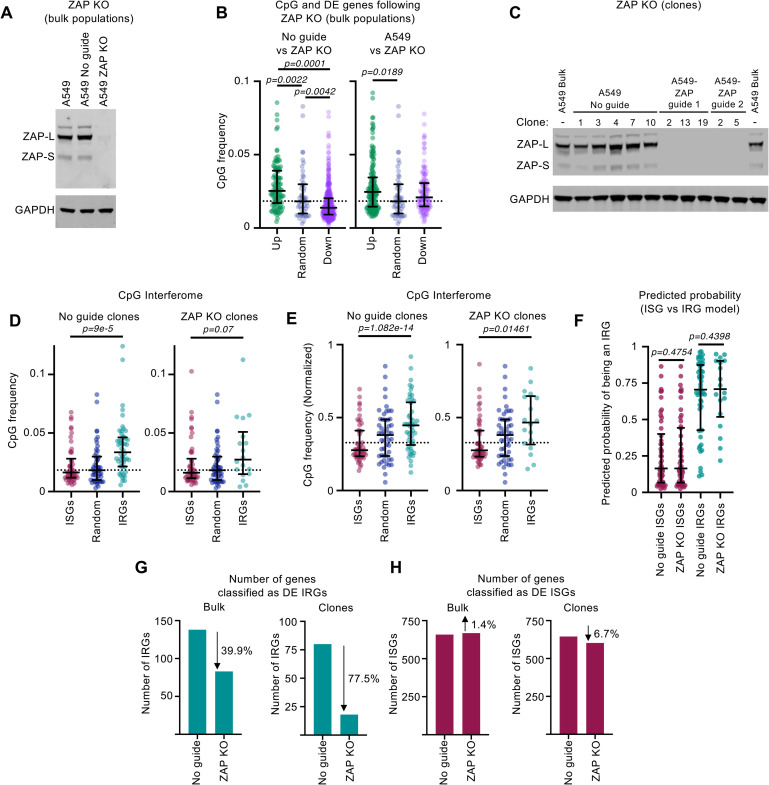
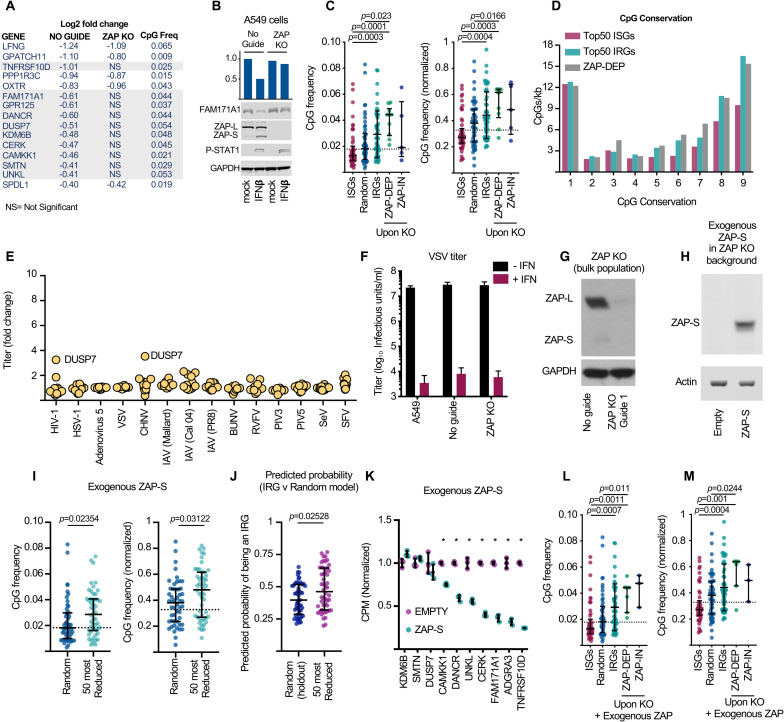
Antiviral defenses can sense viral RNAs and mediate their destruction. This presents a challenge for host cells since they must destroy viral RNAs while sparing the host mRNAs that encode antiviral effectors. Here, we show that highly upregulated interferon-stimulated genes (ISGs), which encode antiviral proteins, have distinctive nucleotide compositions. We propose that self-targeting by antiviral effectors has selected for ISG transcripts that occupy a less self-targeted sequence space. Following interferon (IFN) stimulation, the CpG-targeting antiviral effector zinc-finger antiviral protein (ZAP) reduces the mRNA abundance of multiple host transcripts, providing a mechanistic explanation for the repression of many (but not all) interferon-repressed genes (IRGs). Notably, IRGs tend to be relatively CpG rich. In contrast, highly upregulated ISGs tend to be strongly CpG suppressed. Thus, ZAP is an example of an effector that has not only selected compositional biases in viral genomes but also appears to have notably shaped the composition of host transcripts in the vertebrate interferome.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Comment in
-
Less is more: Biased loss of CpG dinucleotides strengthens antiviral immunity.PLoS Biol. 2021 Sep 8;19(9):e3001353. doi: 10.1371/journal.pbio.3001353. eCollection 2021 Sep. PLoS Biol. 2021. PMID: 34495970 Free PMC article.
References
-
- Shaw AE, Hughes J, Gu Q, Behdenna A, Singer JB, Dennis T, et al. Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol. 2017;15(12). ARTN e2004086.10.1371/journal.pbio.2004086. WOS:000418943900021. doi: 10.1371/journal.pbio.2004086 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
