

A compendium of uniformly processed human gene expression and splicing quantitative trait loci
- PMID: 34493866
- PMCID: PMC8423625
- DOI: 10.1038/s41588-021-00924-w
A compendium of uniformly processed human gene expression and splicing quantitative trait loci
Abstract
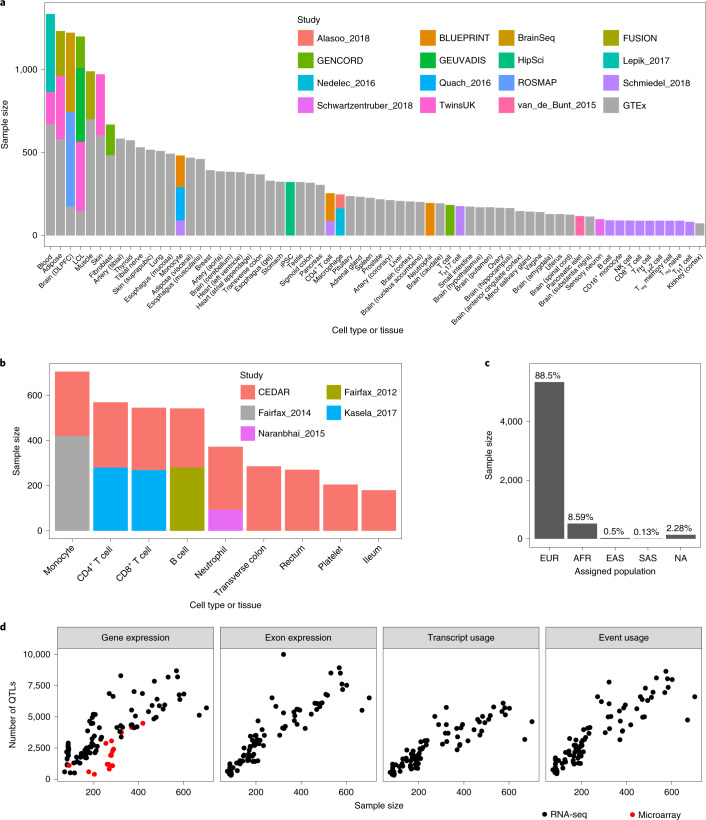
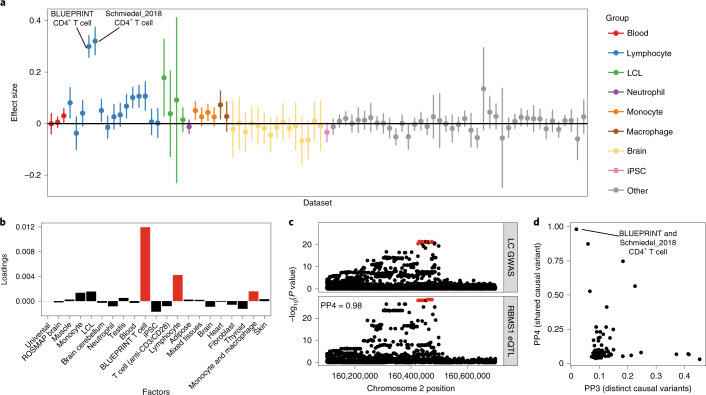
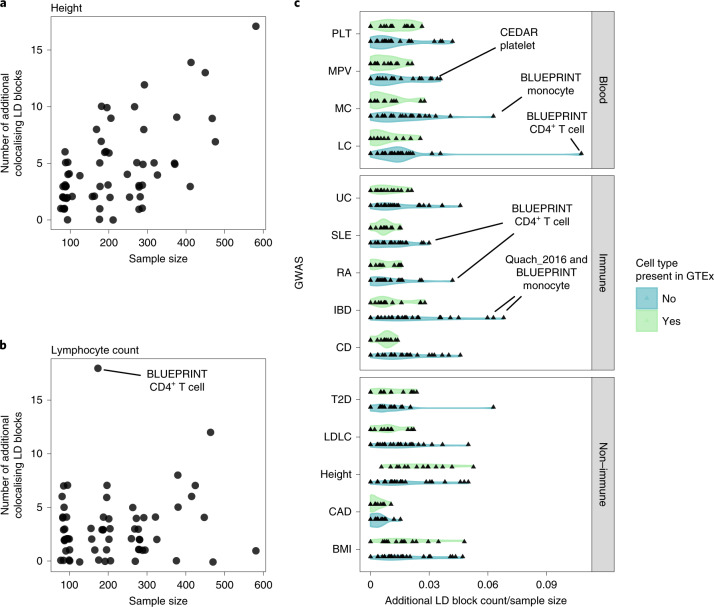
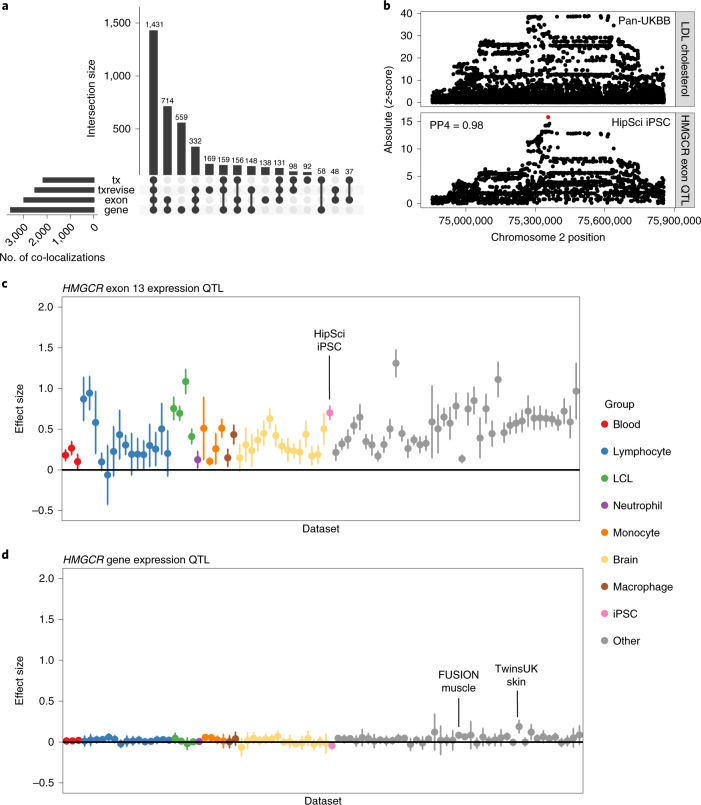
Many gene expression quantitative trait locus (eQTL) studies have published their summary statistics, which can be used to gain insight into complex human traits by downstream analyses, such as fine mapping and co-localization. However, technical differences between these datasets are a barrier to their widespread use. Consequently, target genes for most genome-wide association study (GWAS) signals have still not been identified. In the present study, we present the eQTL Catalogue ( https://www.ebi.ac.uk/eqtl ), a resource of quality-controlled, uniformly re-computed gene expression and splicing QTLs from 21 studies. We find that, for matching cell types and tissues, the eQTL effect sizes are highly reproducible between studies. Although most QTLs were shared between most bulk tissues, we identified a greater diversity of cell-type-specific QTLs from purified cell types, a subset of which also manifested as new disease co-localizations. Our summary statistics are freely available to enable the systematic interpretation of human GWAS associations across many cell types and tissues.
© 2021. The Author(s).
Conflict of interest statement
Since April 2021, D.R.Z. has been a full-time employee of Mosaic Therapeutics, UK. The other authors declare no competing interests.
Figures


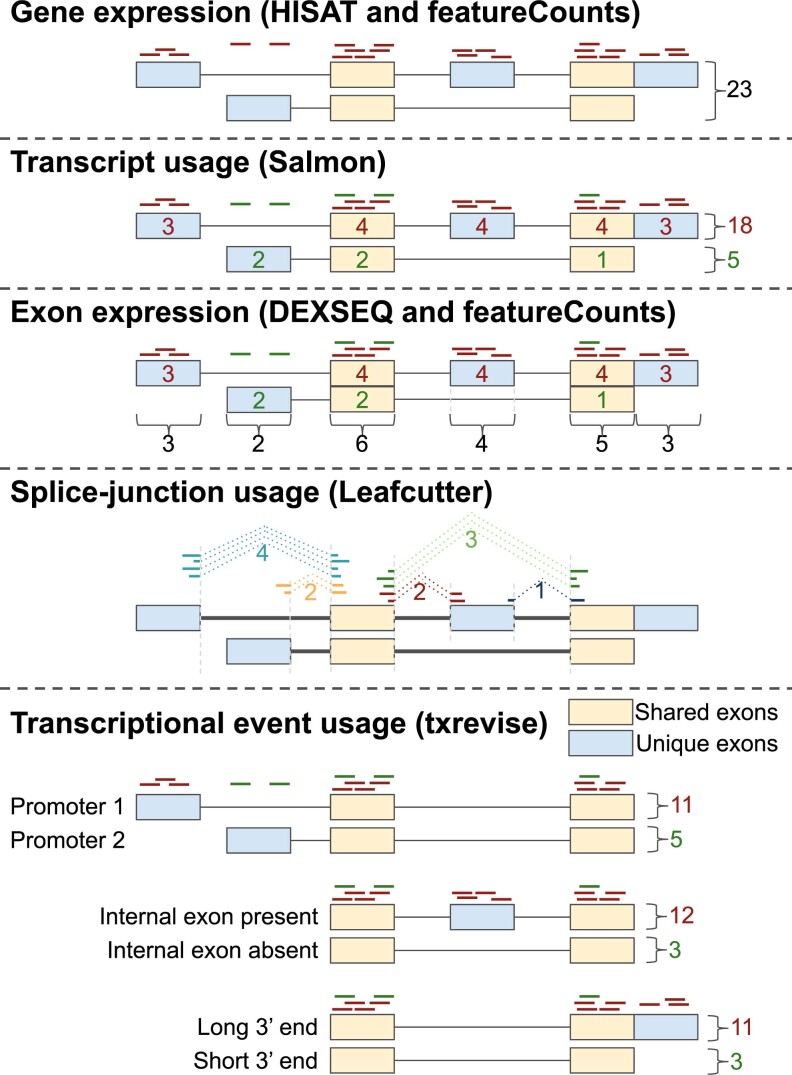



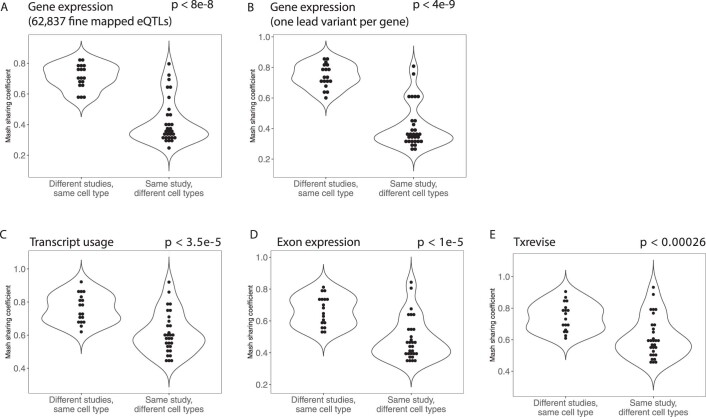
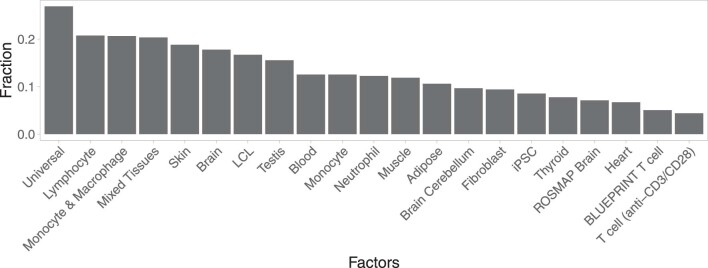
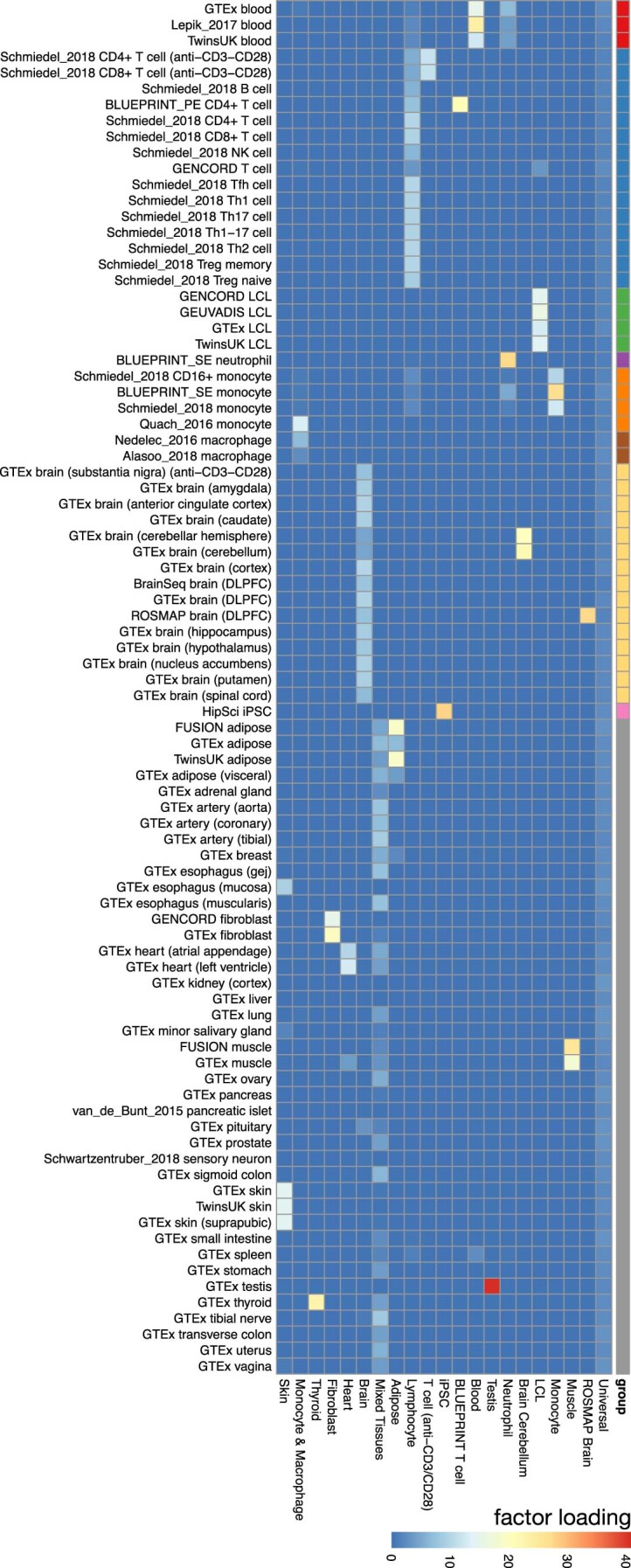


References
-
- Võsa, U. et al. Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis. Preprint at bioRxiv10.1101/447367 (2018).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
