

Genomic and evolutionary classification of lung cancer in never smokers
- PMID: 34493867
- PMCID: PMC8432745
- DOI: 10.1038/s41588-021-00920-0
Genomic and evolutionary classification of lung cancer in never smokers
Abstract
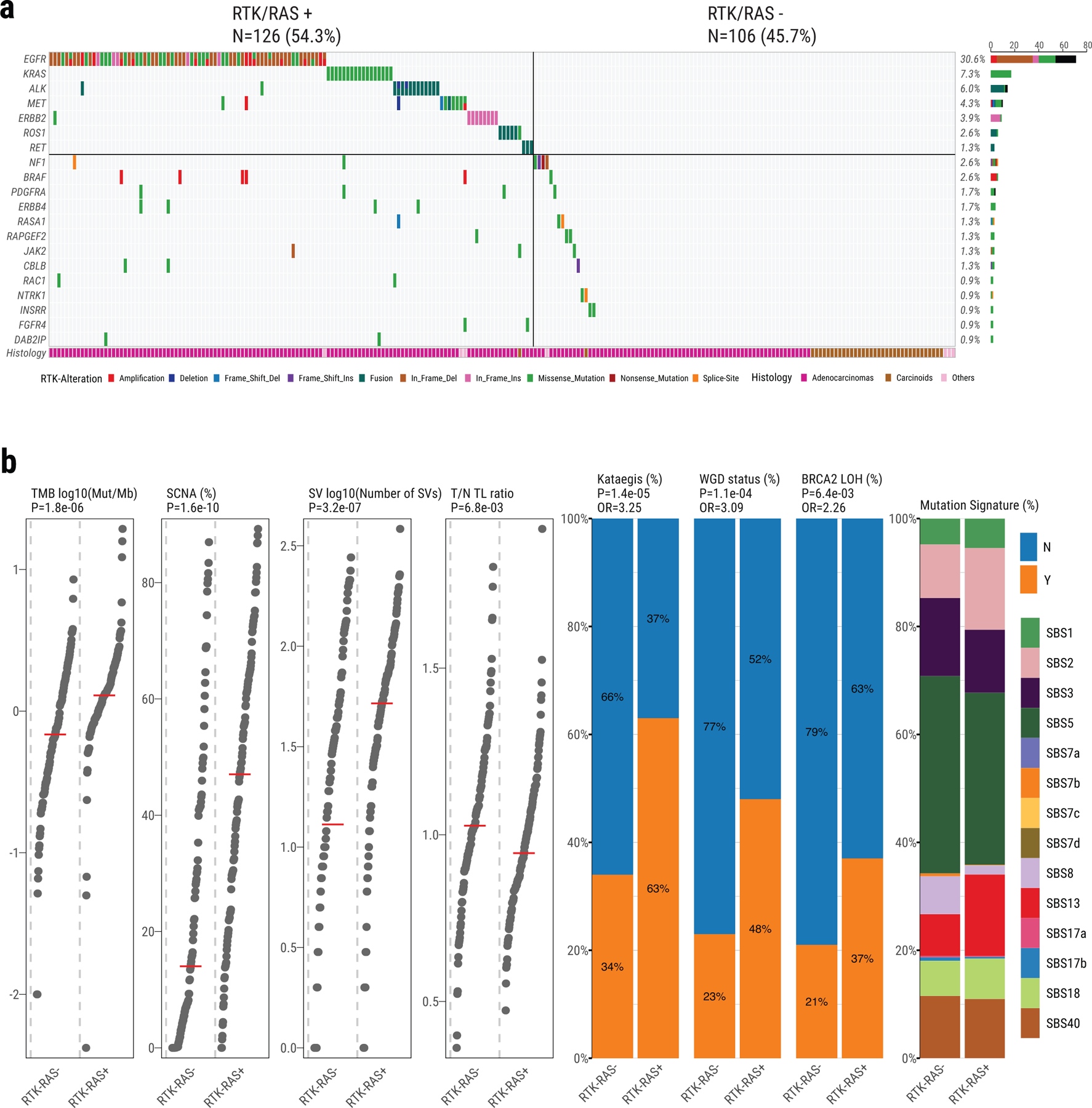
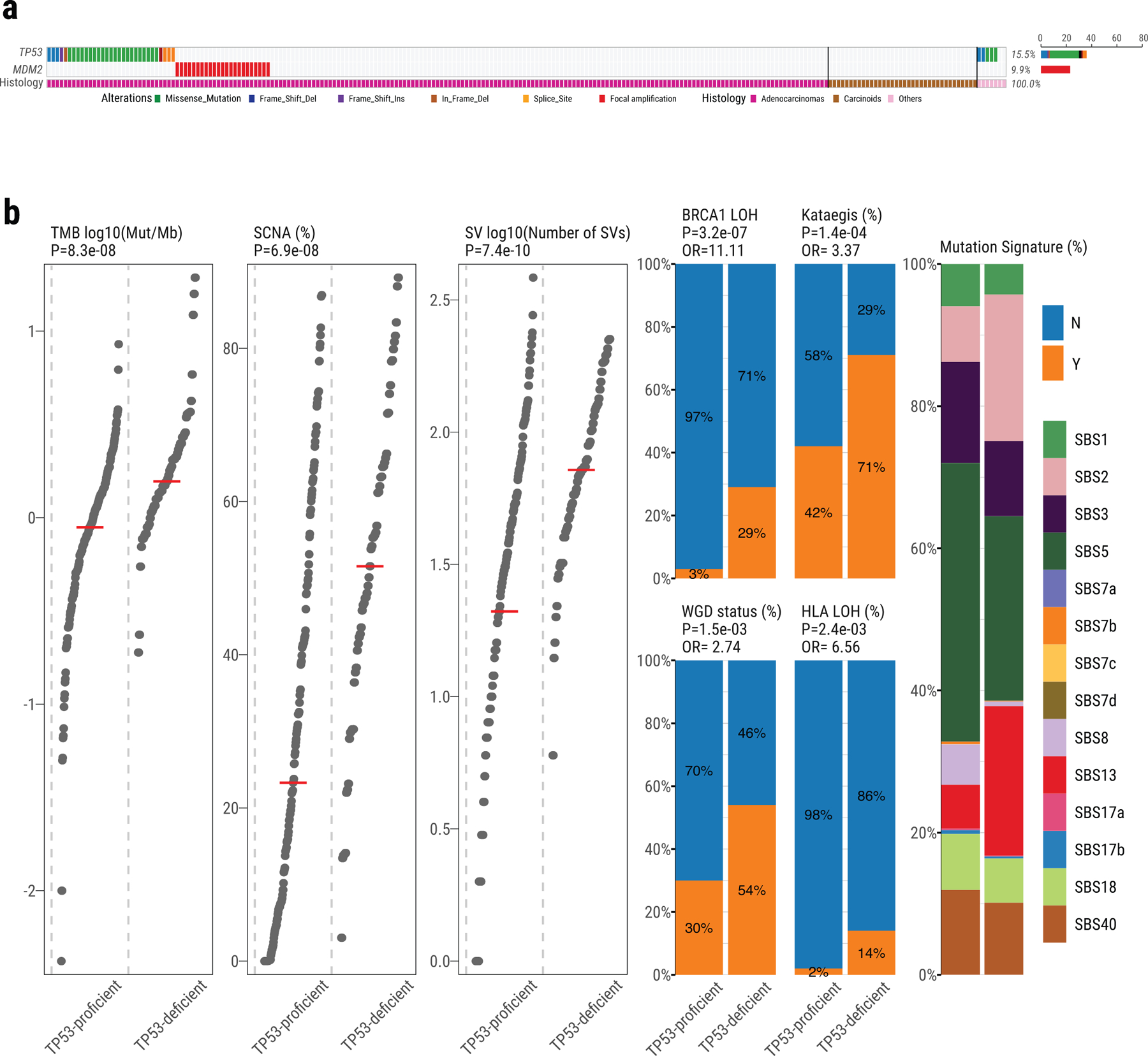
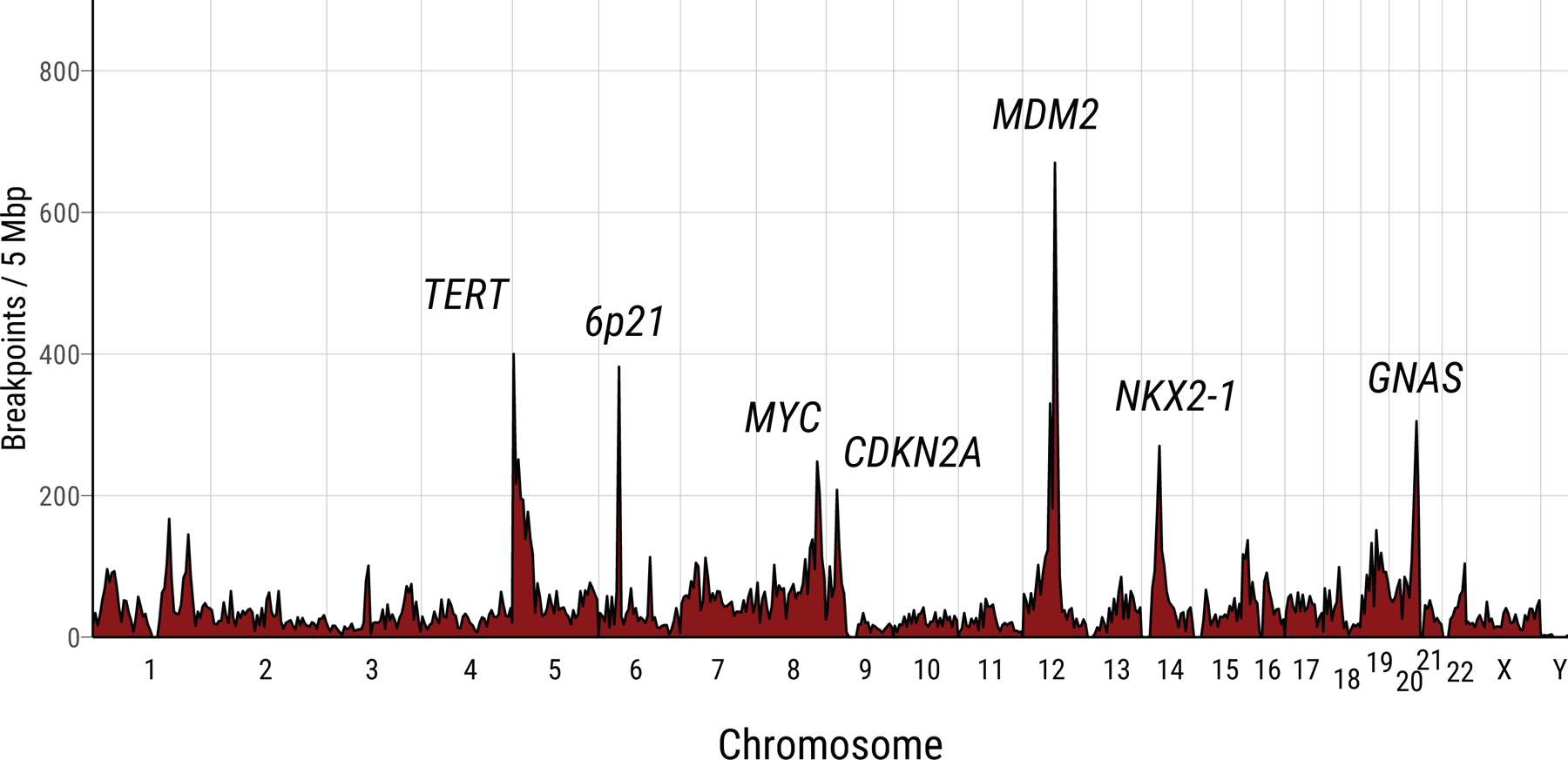
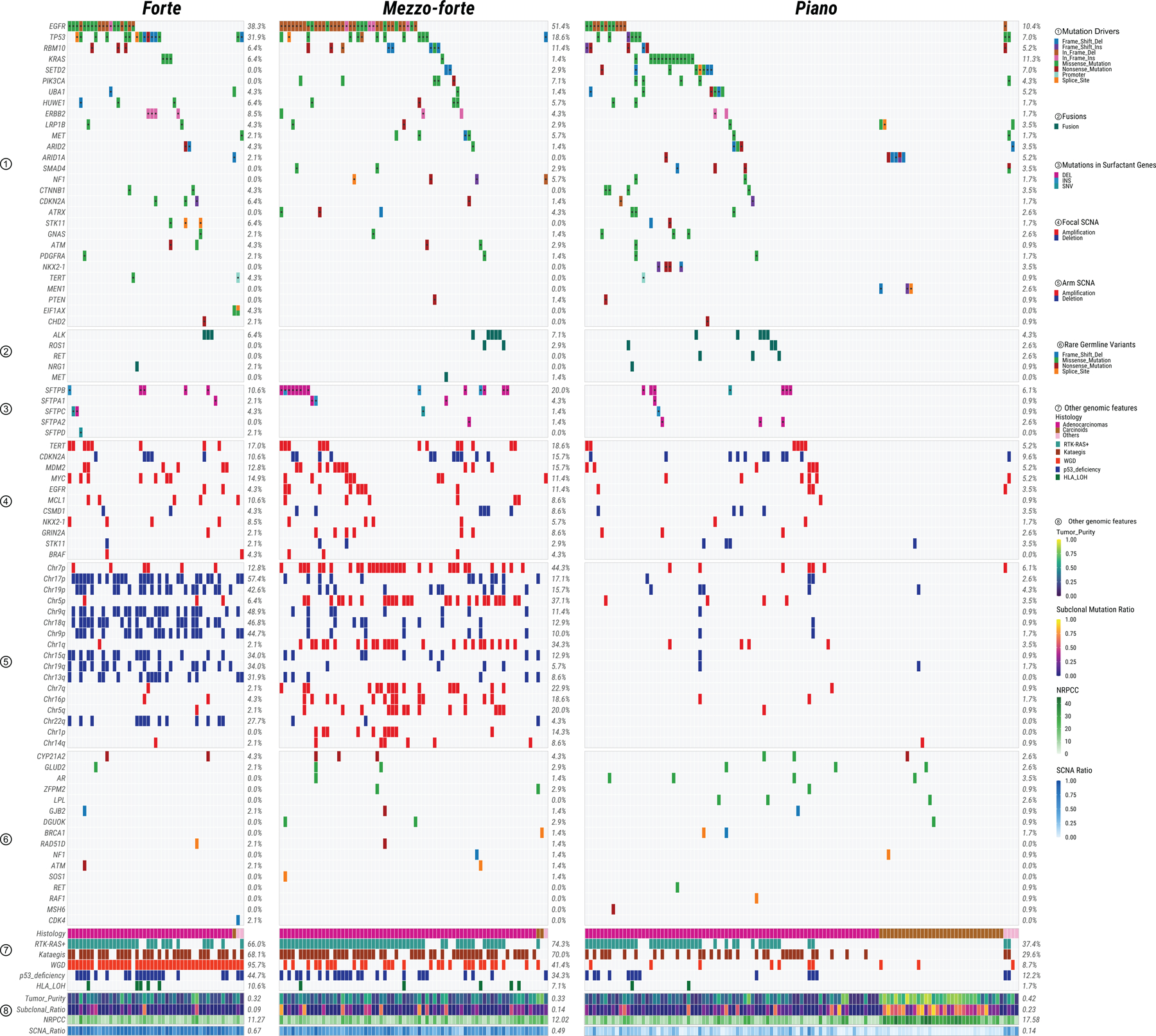
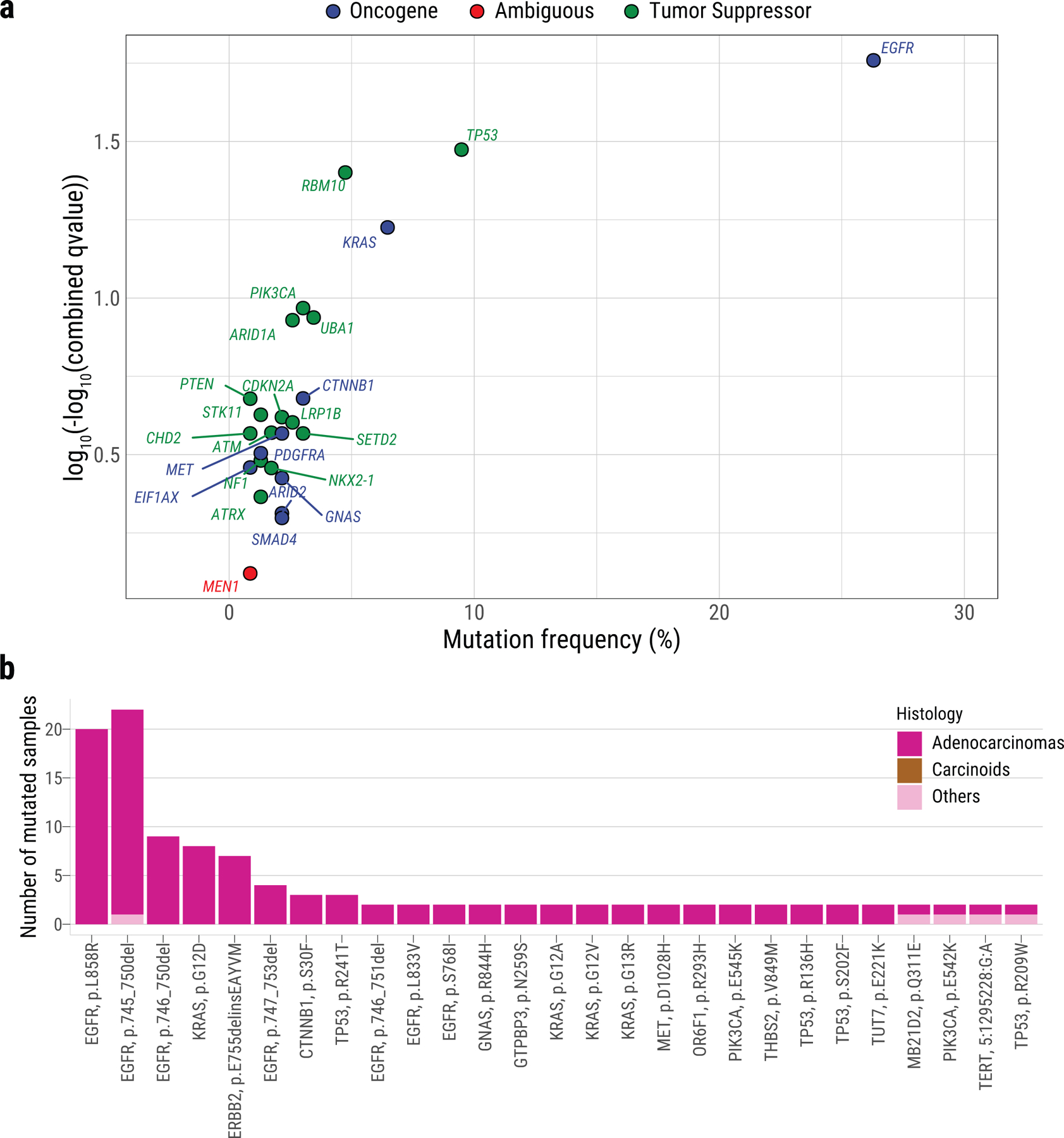
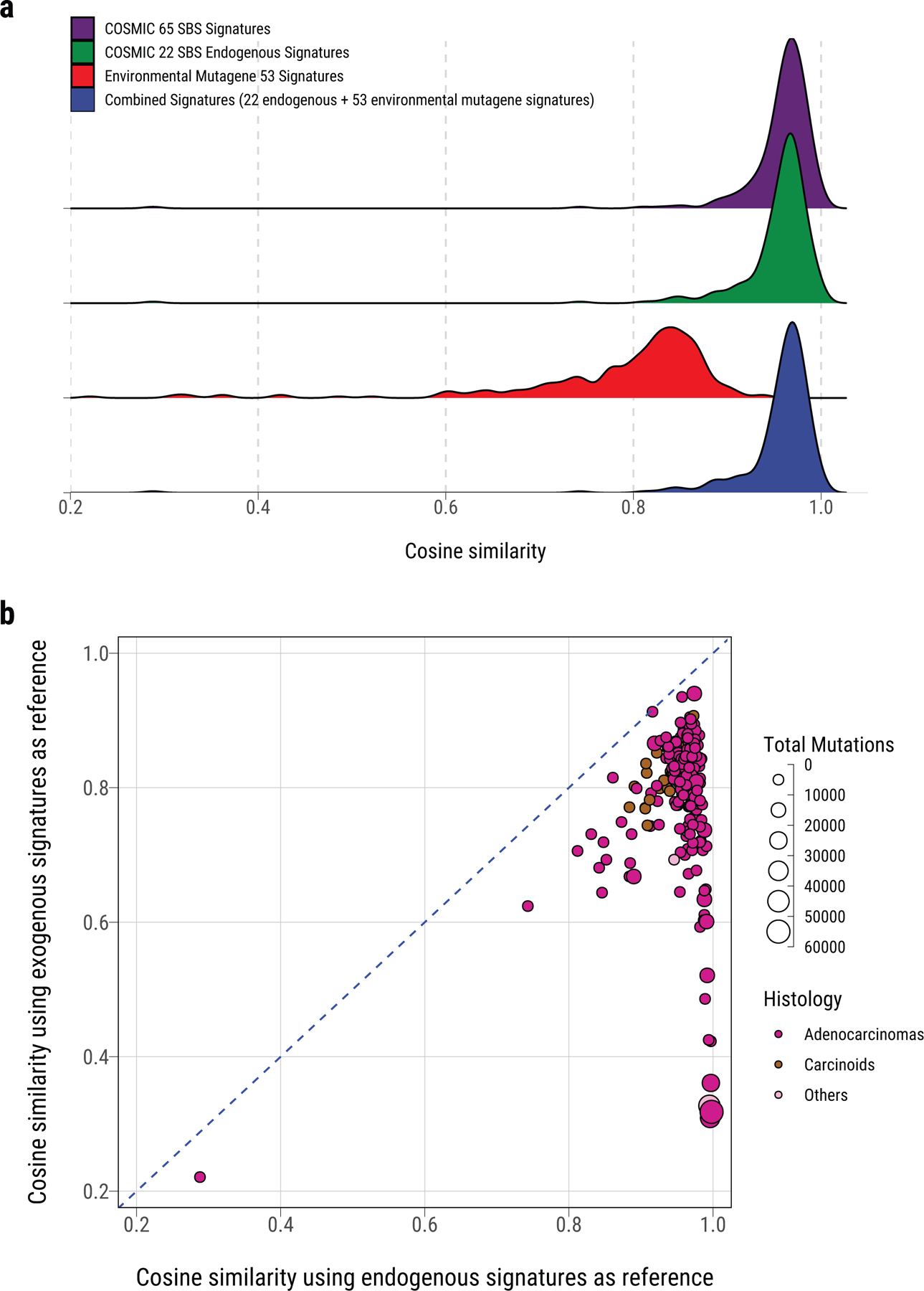
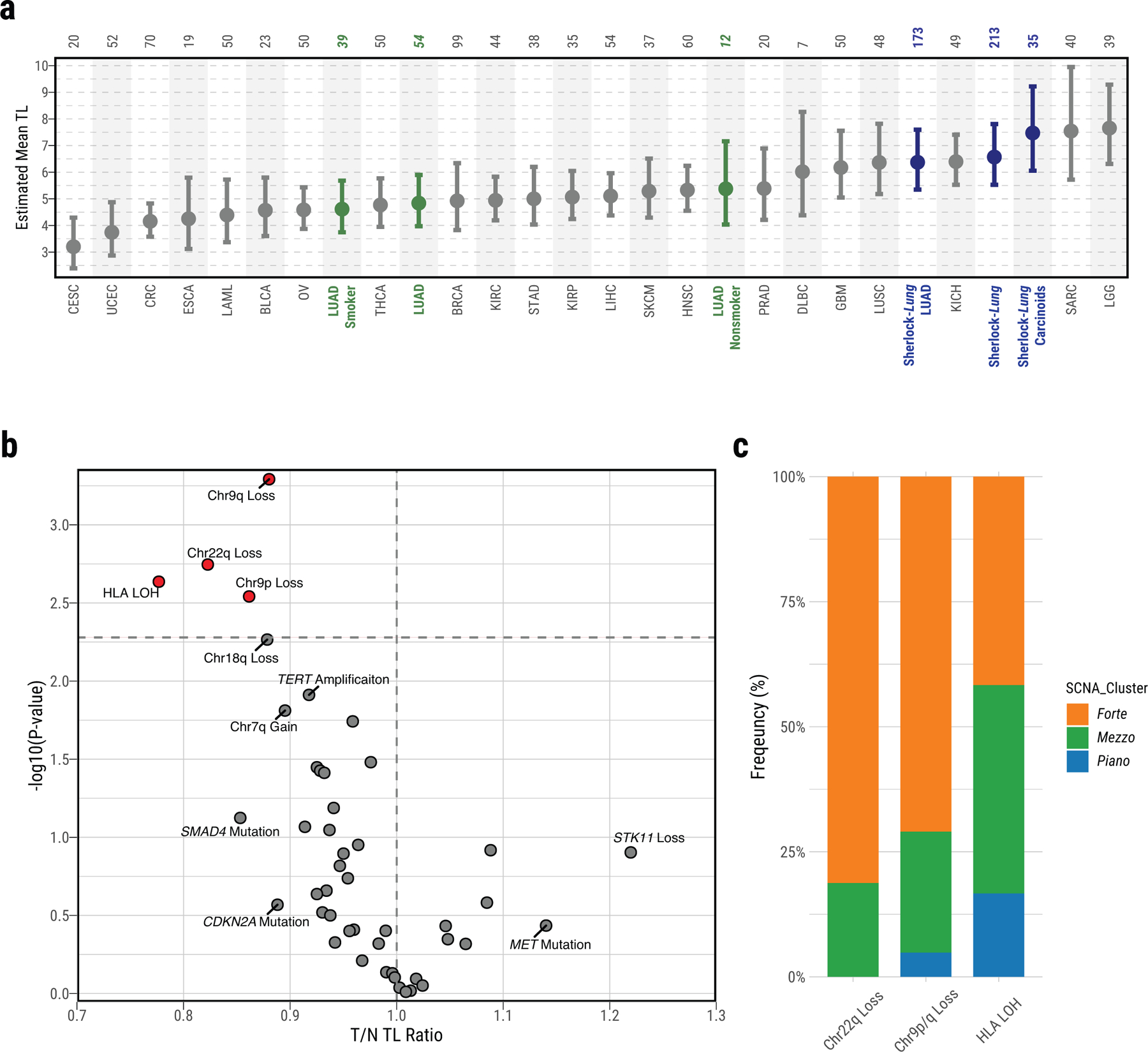
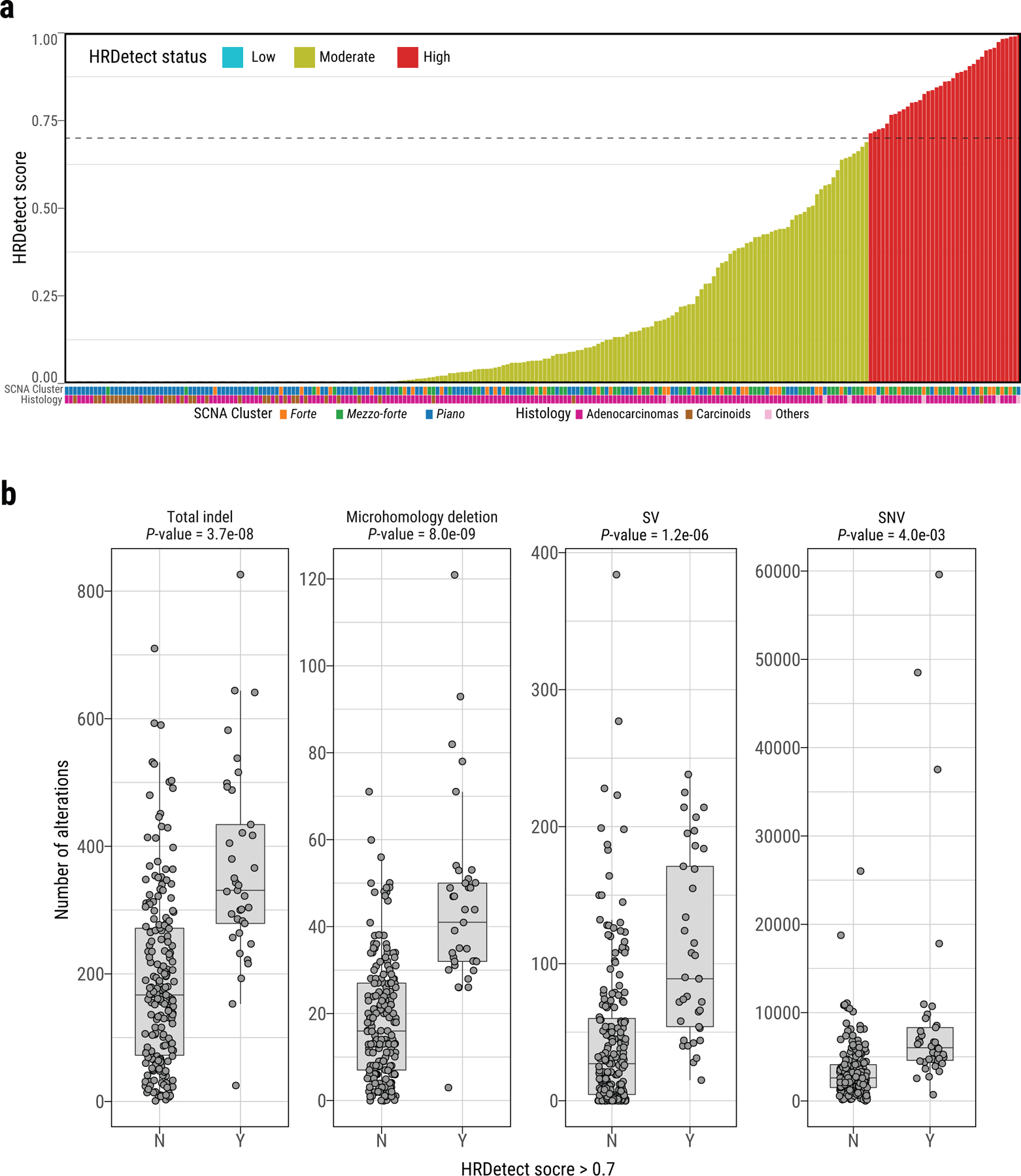
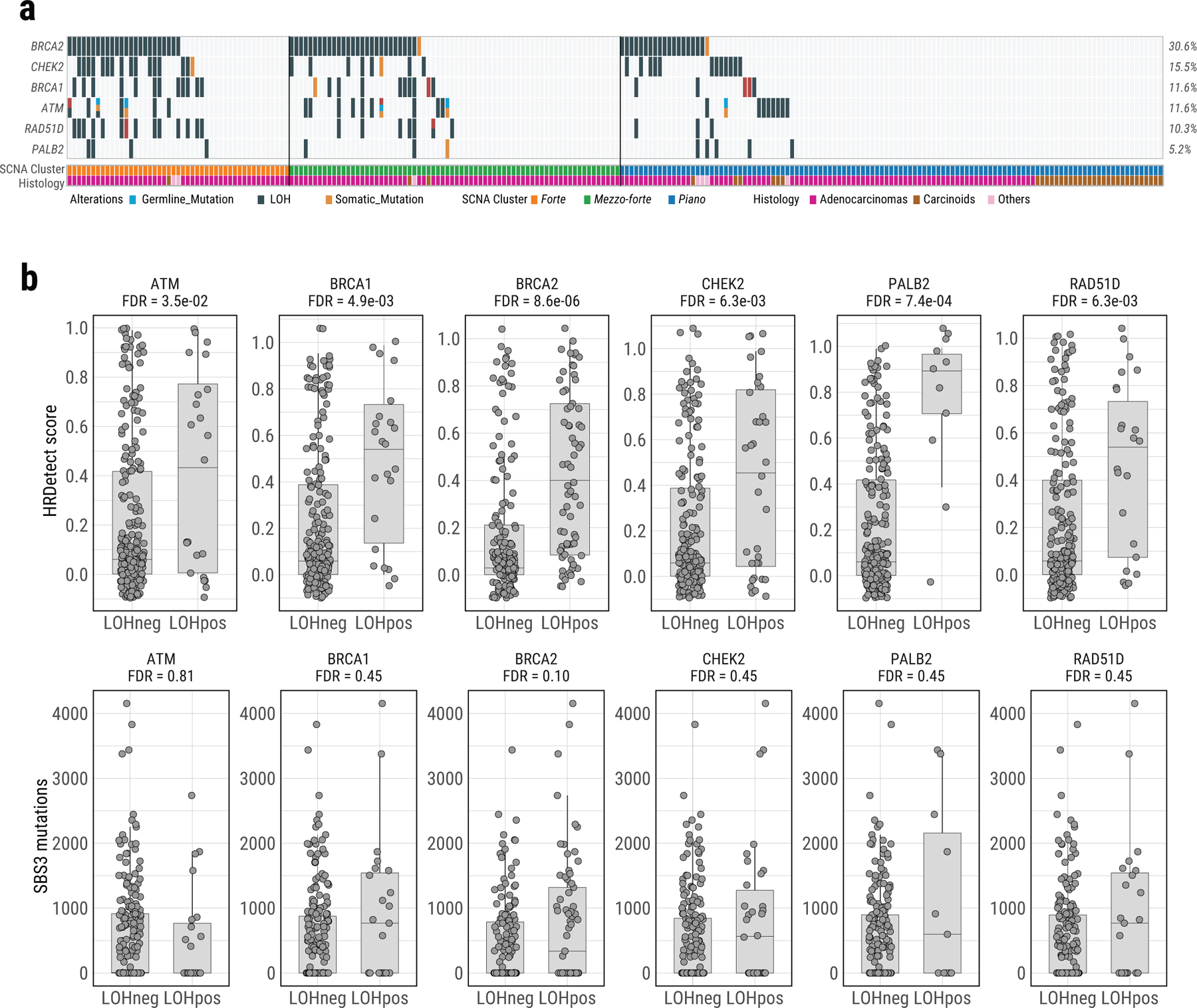
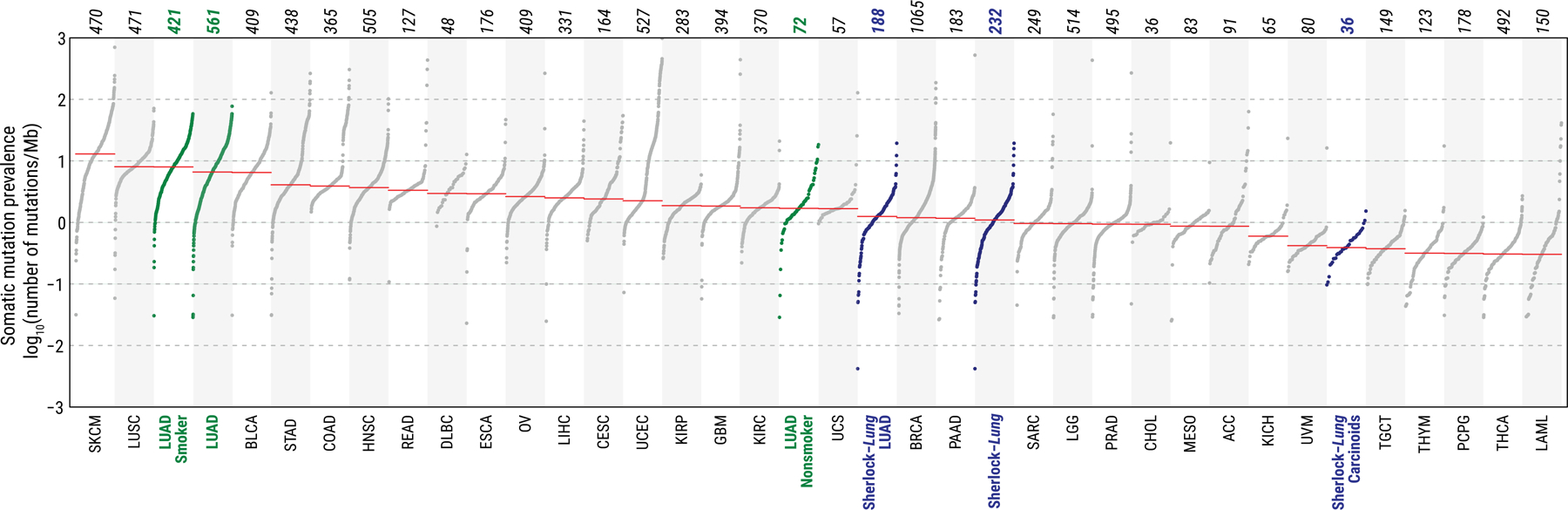
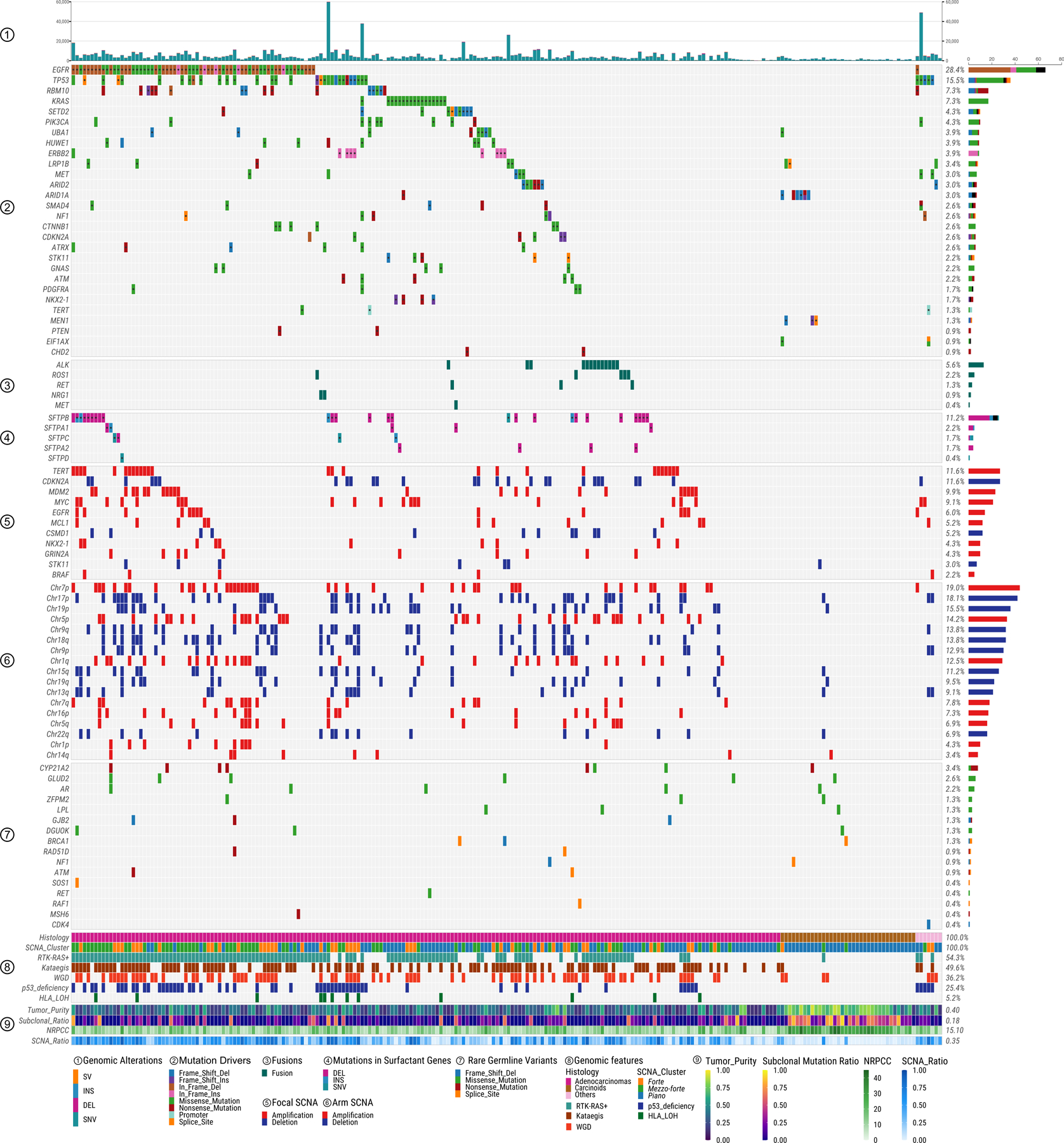
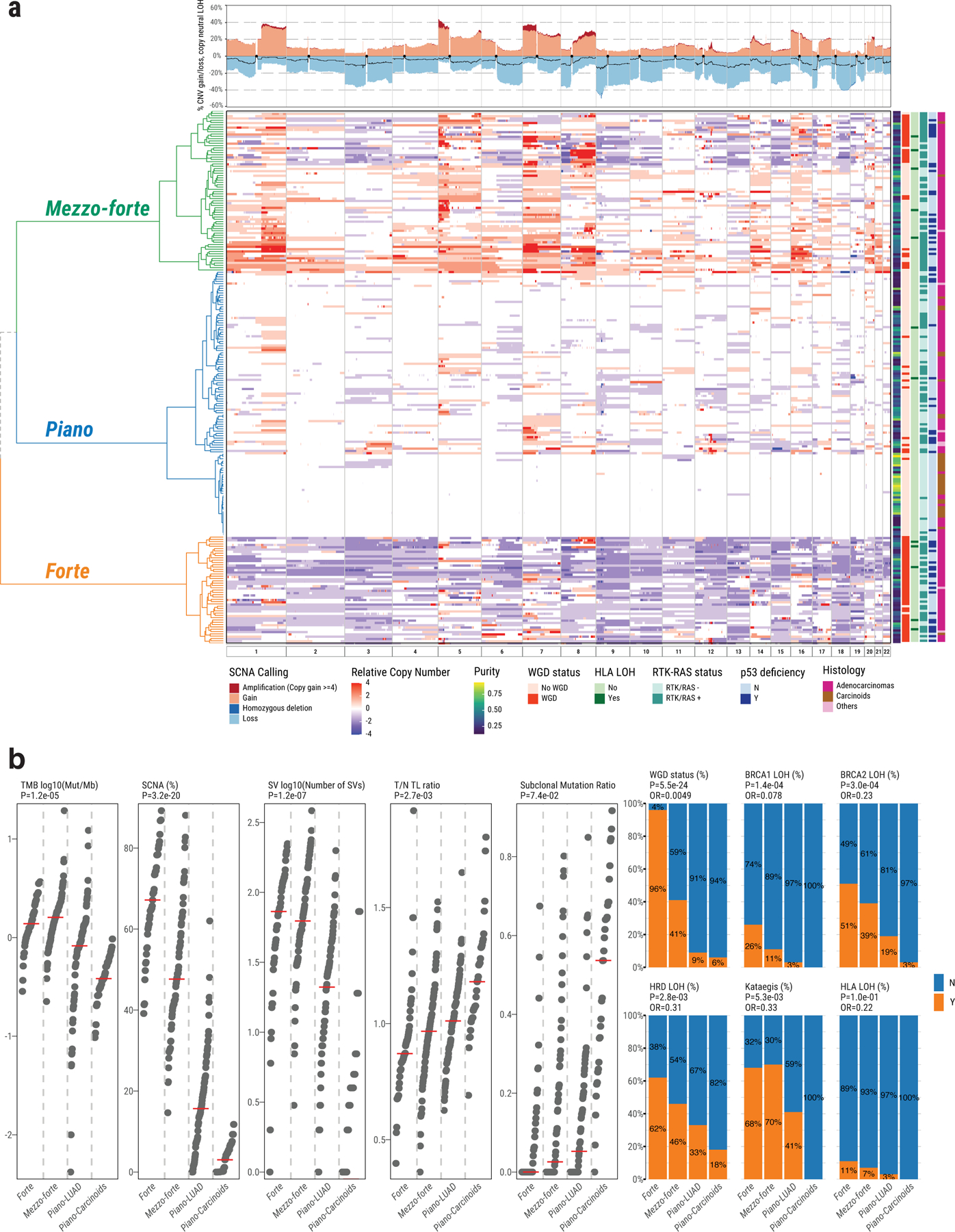
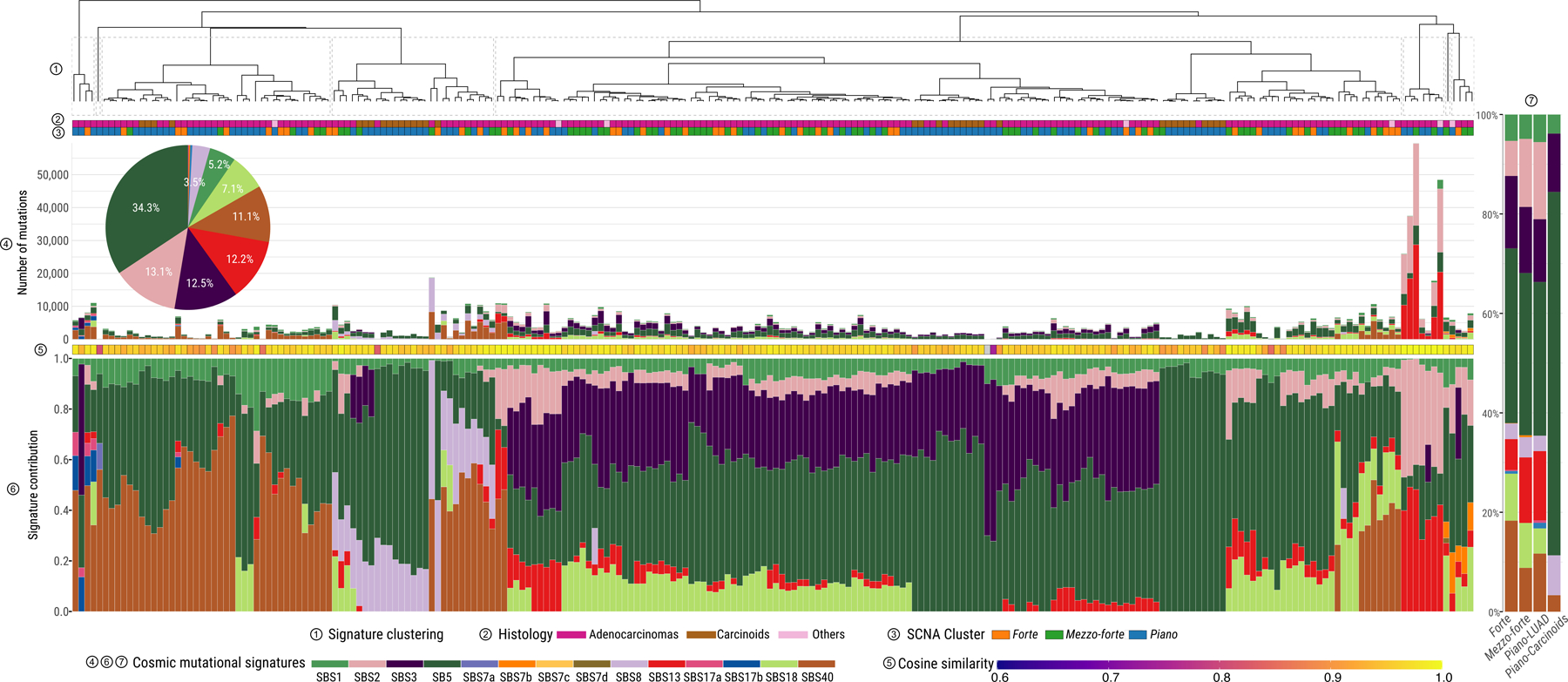
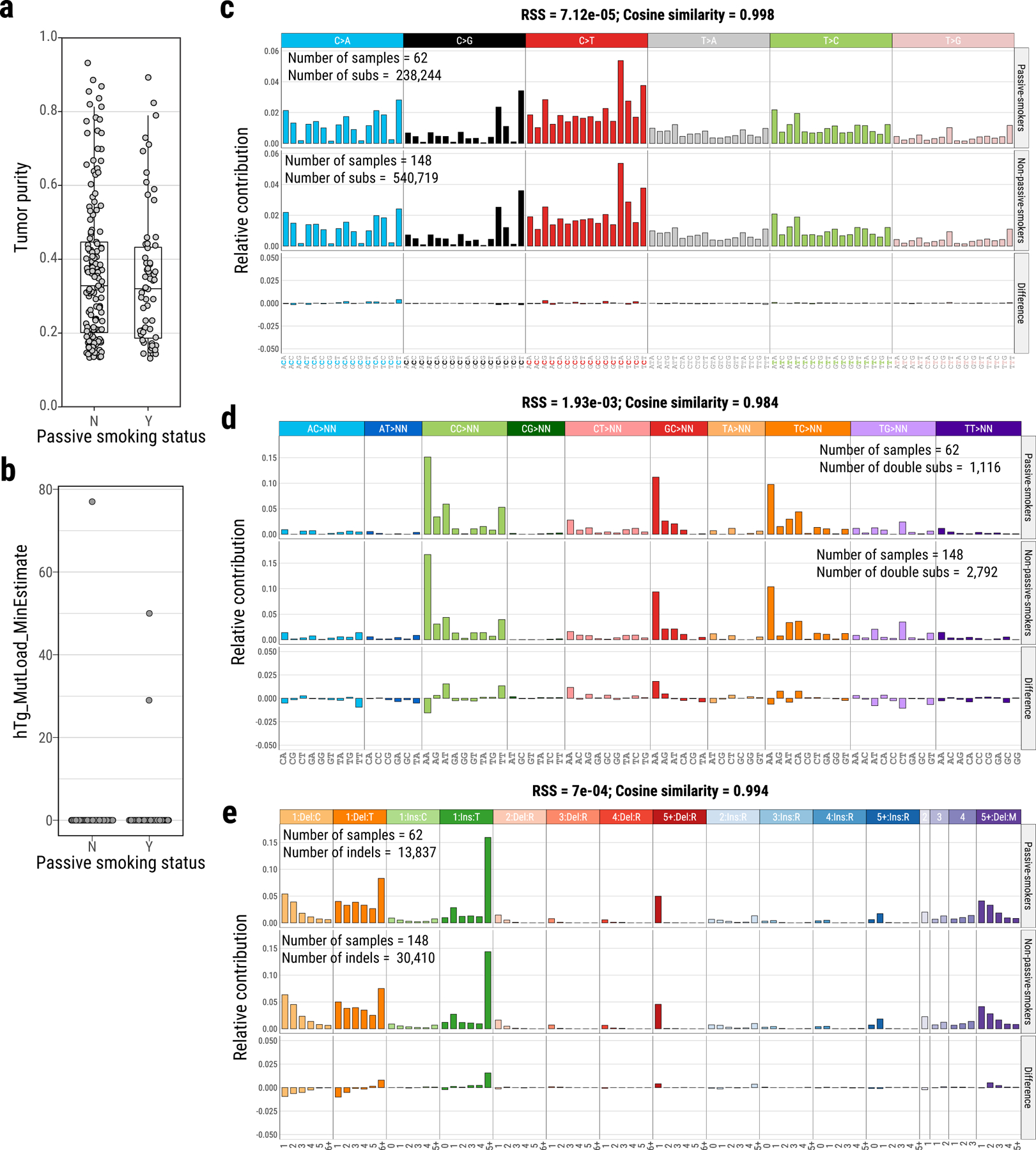
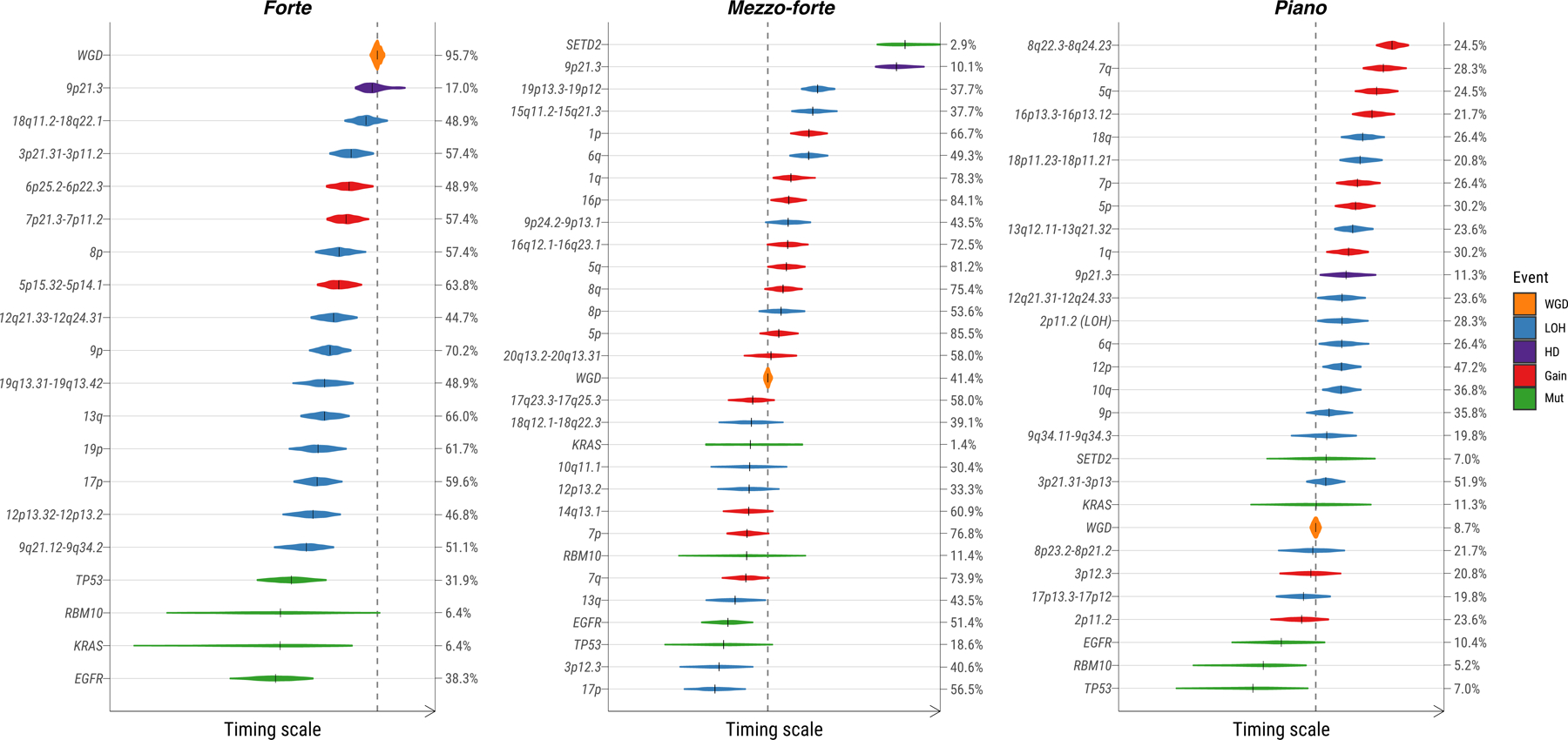
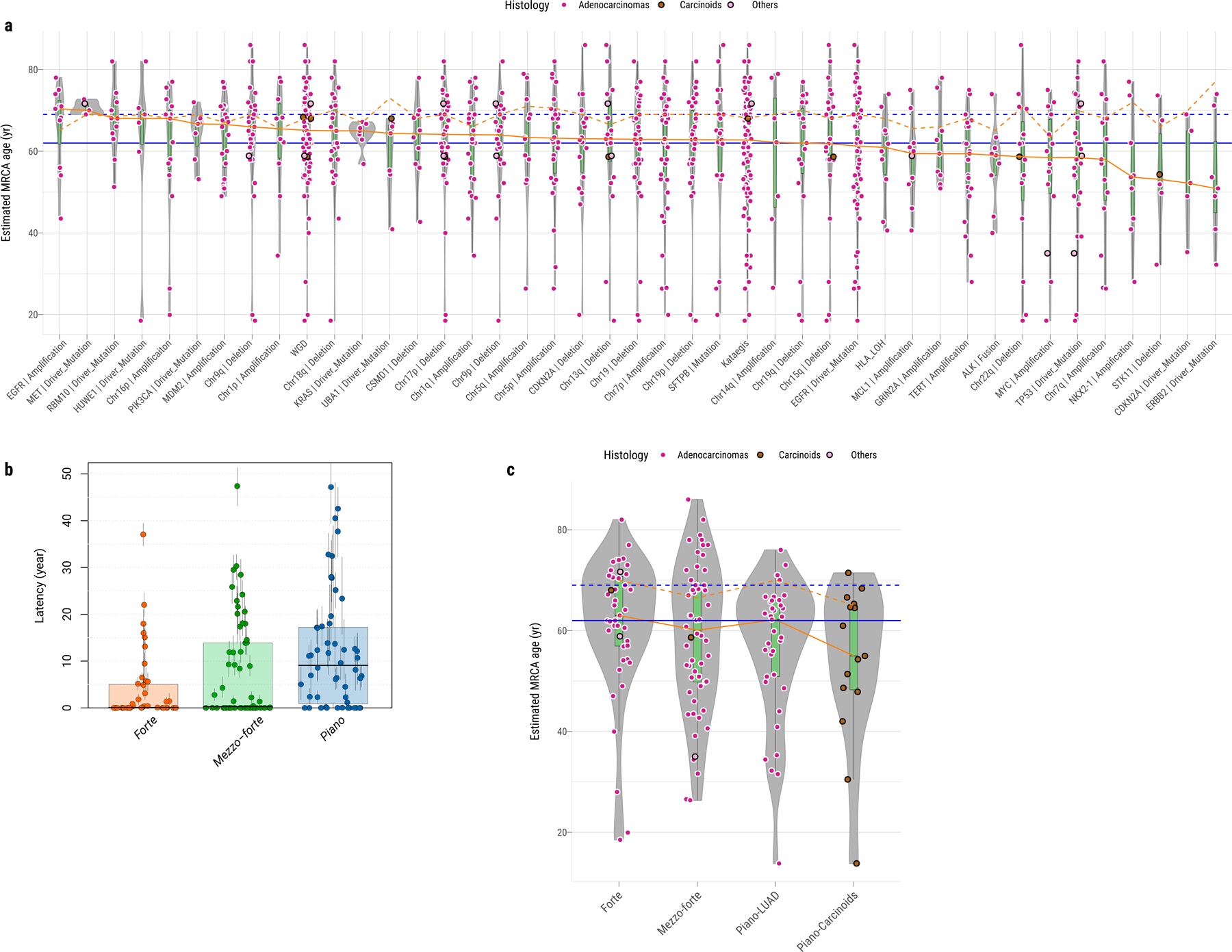
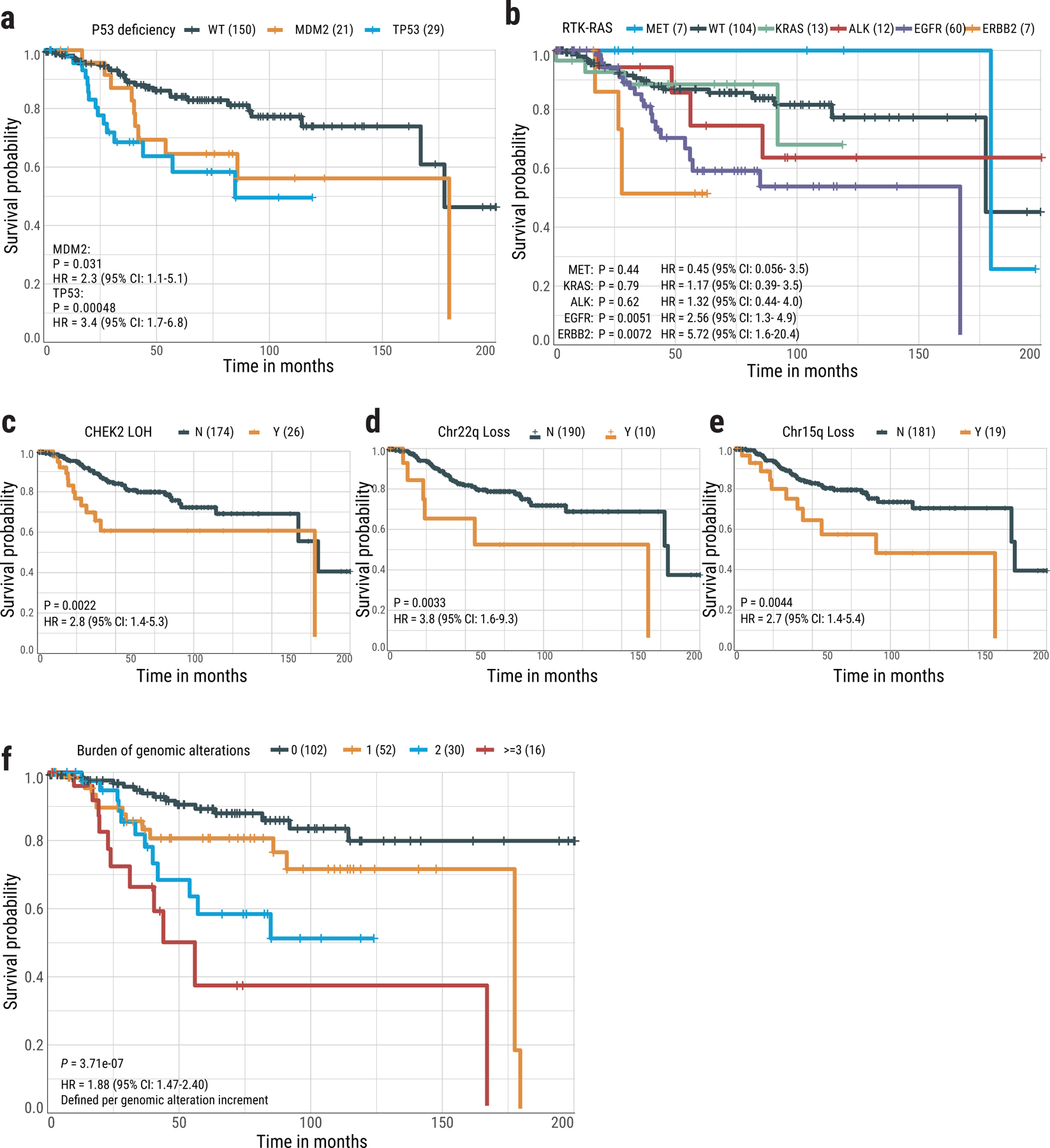
Lung cancer in never smokers (LCINS) is a common cause of cancer mortality but its genomic landscape is poorly characterized. Here high-coverage whole-genome sequencing of 232 LCINS showed 3 subtypes defined by copy number aberrations. The dominant subtype (piano), which is rare in lung cancer in smokers, features somatic UBA1 mutations, germline AR variants and stem cell-like properties, including low mutational burden, high intratumor heterogeneity, long telomeres, frequent KRAS mutations and slow growth, as suggested by the occurrence of cancer drivers' progenitor cells many years before tumor diagnosis. The other subtypes are characterized by specific amplifications and EGFR mutations (mezzo-forte) and whole-genome doubling (forte). No strong tobacco smoking signatures were detected, even in cases with exposure to secondhand tobacco smoke. Genes within the receptor tyrosine kinase-Ras pathway had distinct impacts on survival; five genomic alterations independently doubled mortality. These findings create avenues for personalized treatment in LCINS.
© 2021. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
Comment in
-
Developments in lung cancer biology in never-smokers.Lancet Oncol. 2021 Oct;22(10):1363. doi: 10.1016/S1470-2045(21)00532-5. Epub 2021 Sep 9. Lancet Oncol. 2021. PMID: 34509183 No abstract available.
References
-
- The Cancer Atlas: Lung Cancer. The Cancer Atlas https://canceratlas.cancer.org/the-burden/lung-cancer/.
-
- Chen J. et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet 52, 177–186 (2020). - PubMed
Method References
-
- Jørsboe E, Hanghøj K & Albrechtsen A fastNGSadmix: admixture proportions and principal component analysis of a single NGS sample. Bioinformatics 33, 3148–3150 (2017). - PubMed
-
- Kim S. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018). - PubMed
-
- Freed D, Pan R & Aldana R TNscope: Accurate Detection of Somatic Mutations with Haplotype-based Variant Candidate Detection and Machine Learning Filtering. bioRxiv 250647 (2018) doi: 10.1101/250647. - DOI
Publication types
MeSH terms
Substances
Grants and funding
- 75N91019D00024/CA/NCI NIH HHS/United States
- 203141/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- R01 ES032547/ES/NIEHS NIH HHS/United States
- Z01 ES050159/ImNIH/Intramural NIH HHS/United States
- P30 CA076292/CA/NCI NIH HHS/United States
- K08 CA151645/CA/NCI NIH HHS/United States
- R35 HL150876/HL/NHLBI NIH HHS/United States
- Z99 CA999999/ImNIH/Intramural NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- HHSN261201800001C/CA/NCI NIH HHS/United States
- ZIA CP101231/ImNIH/Intramural NIH HHS/United States
- ZIA ES103266/ImNIH/Intramural NIH HHS/United States
- P50 CA196530/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
