

Nonredundant, isoform-specific roles of HDAC1 in glioma stem cells
- PMID: 34494550
- PMCID: PMC8492336
- DOI: 10.1172/jci.insight.149232
Nonredundant, isoform-specific roles of HDAC1 in glioma stem cells
Abstract
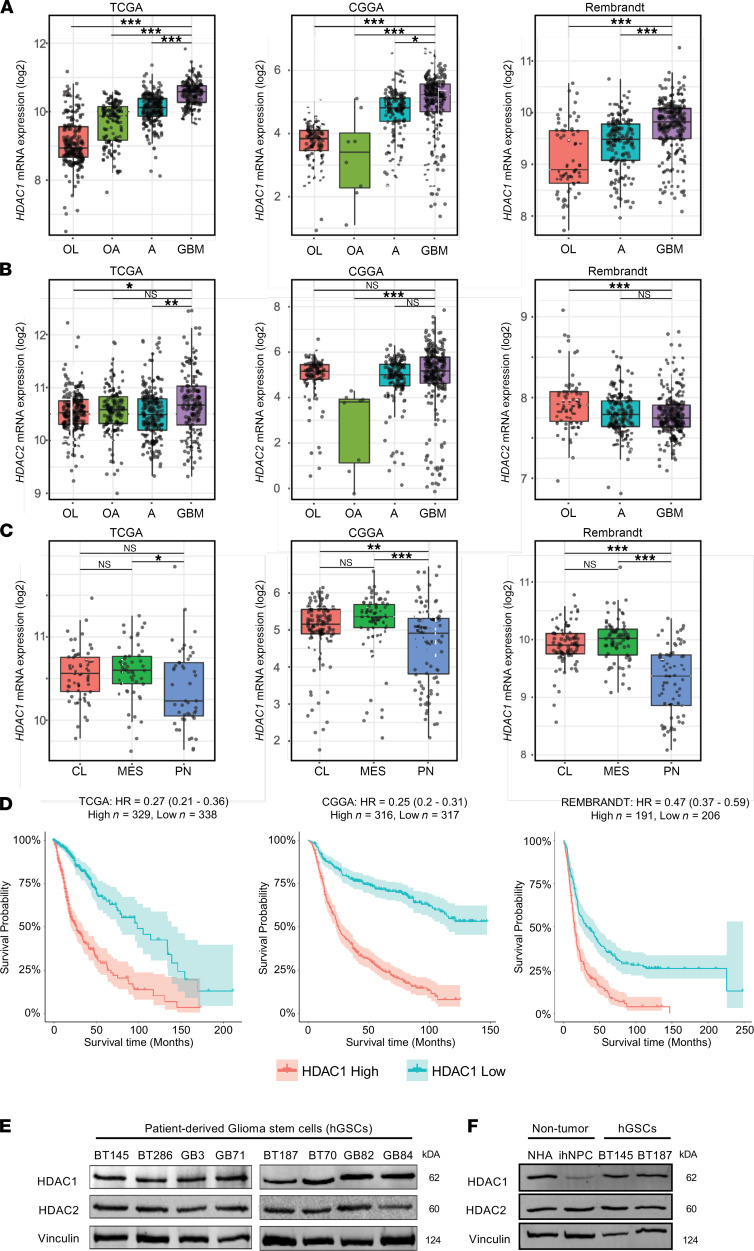
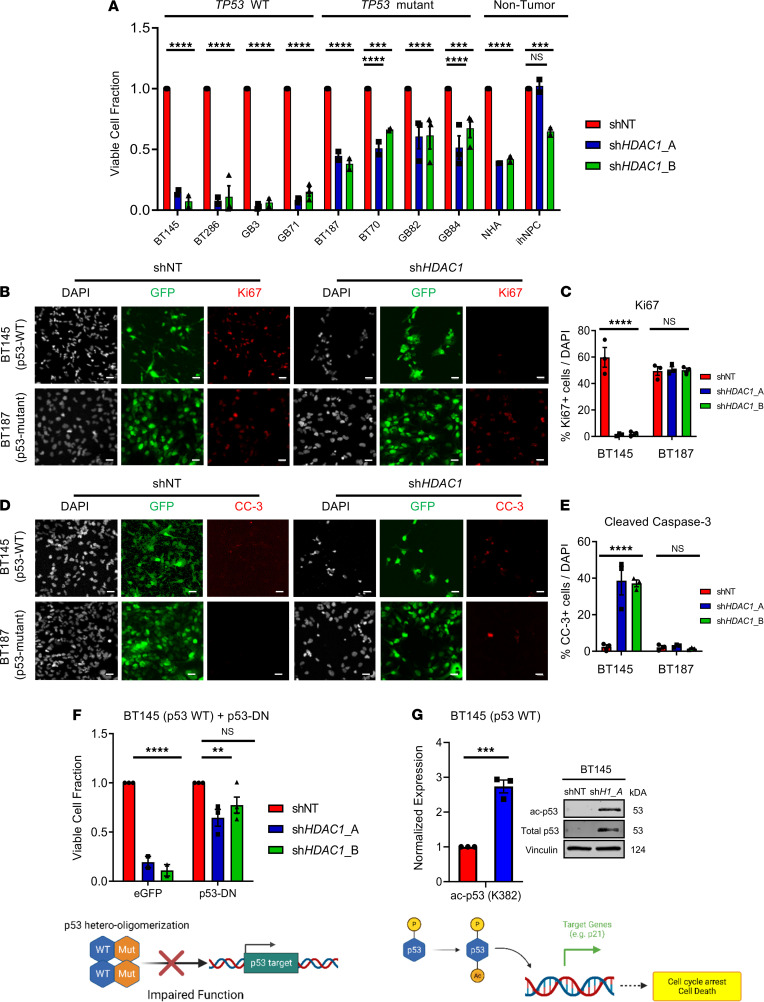
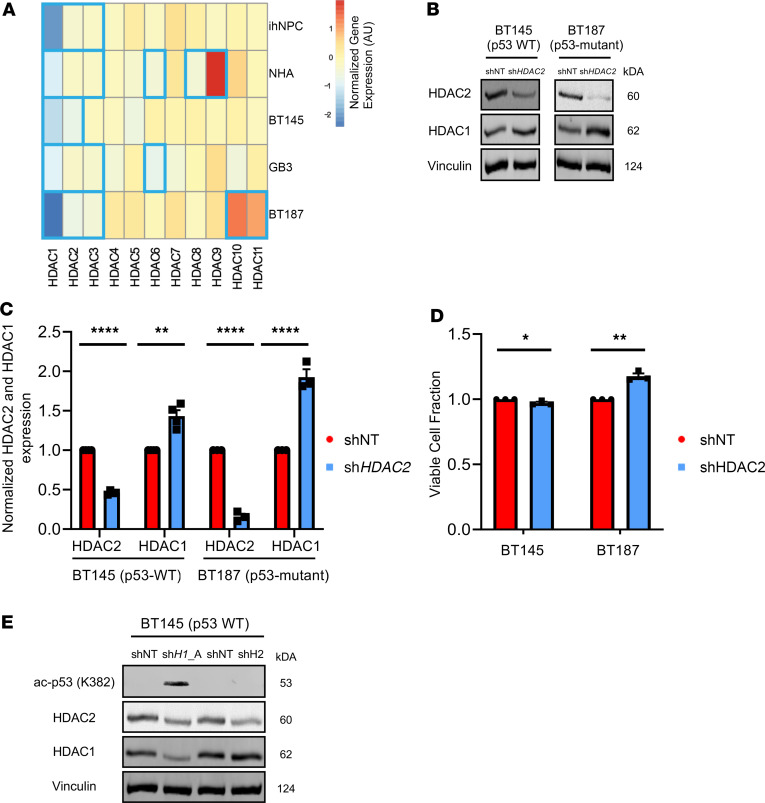
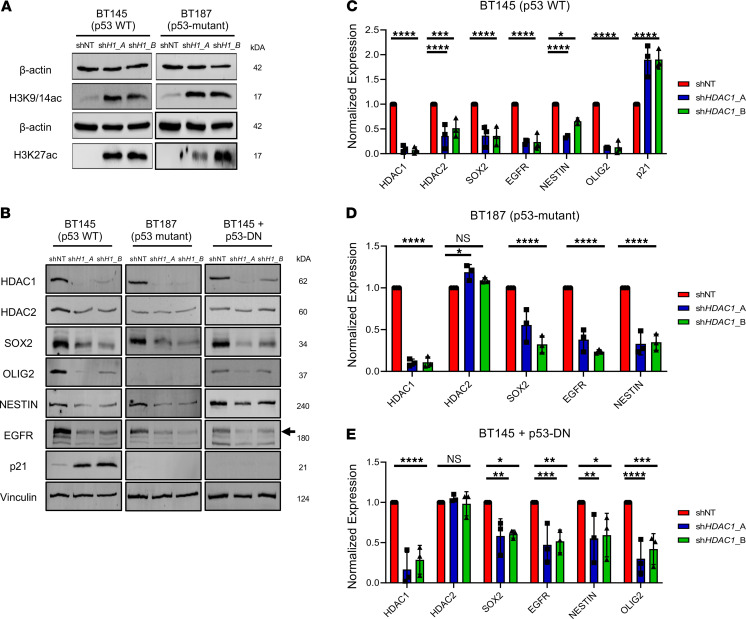
Glioblastoma (GBM) is characterized by an aberrant yet druggable epigenetic landscape. One major family of epigenetic regulators, the histone deacetylases (HDACs), are considered promising therapeutic targets for GBM due to their repressive influences on transcription. Although HDACs share redundant functions and common substrates, the unique isoform-specific roles of different HDACs in GBM remain unclear. In neural stem cells, HDAC2 is the indispensable deacetylase to ensure normal brain development and survival in the absence of HDAC1. Surprisingly, we find that HDAC1 is the essential class I deacetylase in glioma stem cells, and its loss is not compensated for by HDAC2. Using cell-based and biochemical assays, transcriptomic analyses, and patient-derived xenograft models, we find that knockdown of HDAC1 alone has profound effects on the glioma stem cell phenotype in a p53-dependent manner. We demonstrate marked suppression in tumor growth upon targeting of HDAC1 and identify compensatory pathways that provide insights into combination therapies for GBM. Our study highlights the importance of HDAC1 in GBM and the need to develop isoform-specific drugs.
Keywords: Brain cancer; Cancer; Epigenetics; Oncology; Stem cells.
Conflict of interest statement
Figures
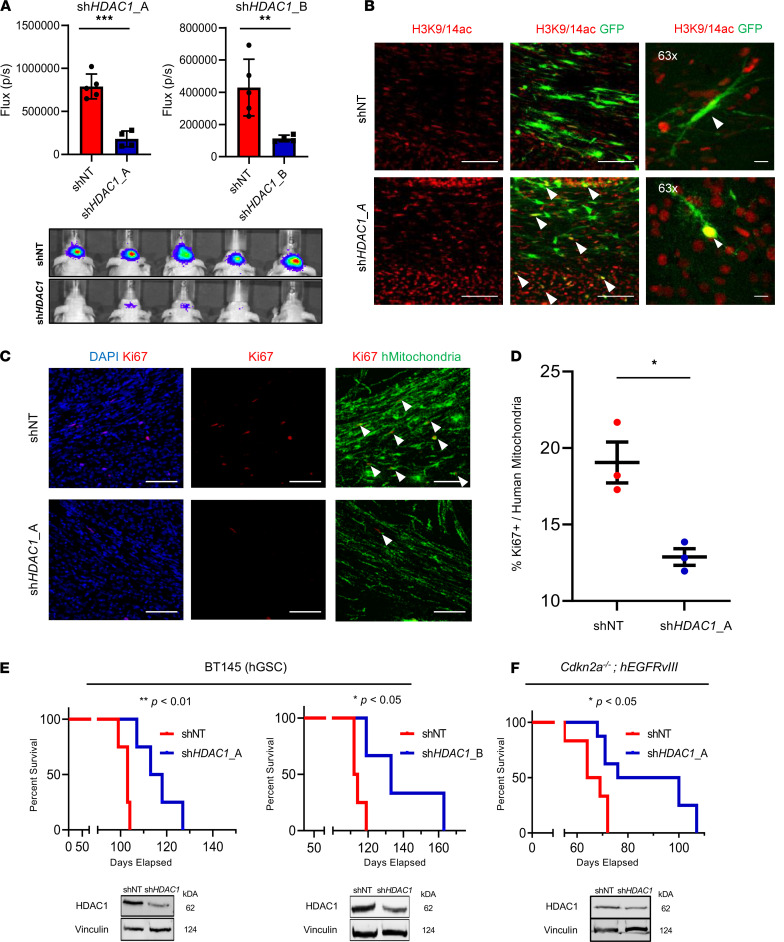
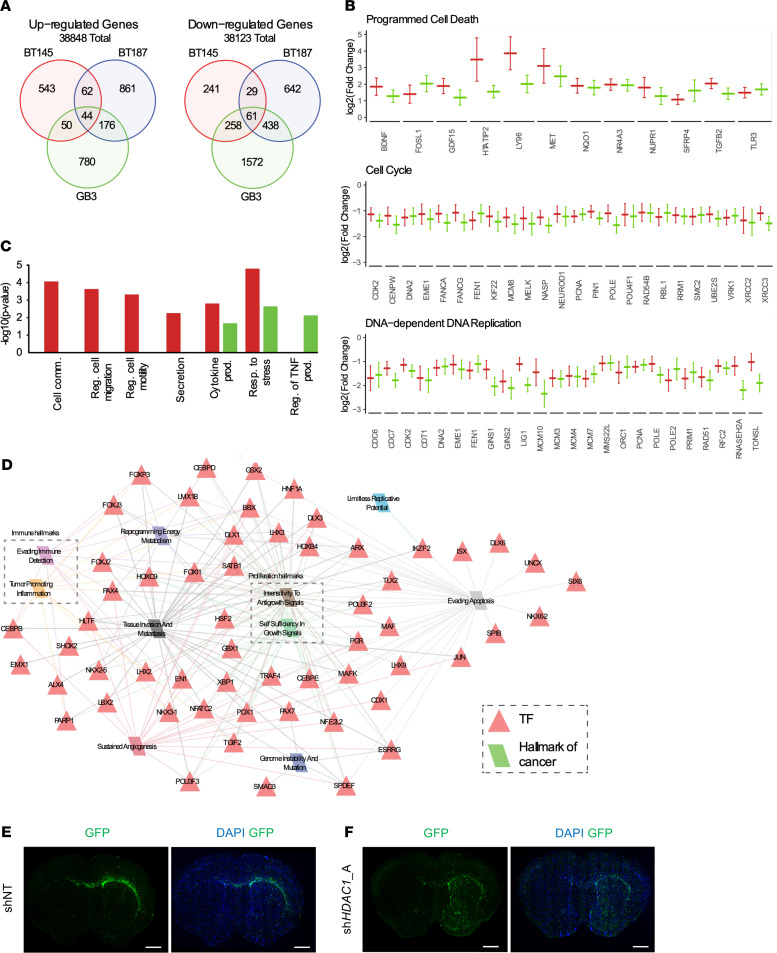
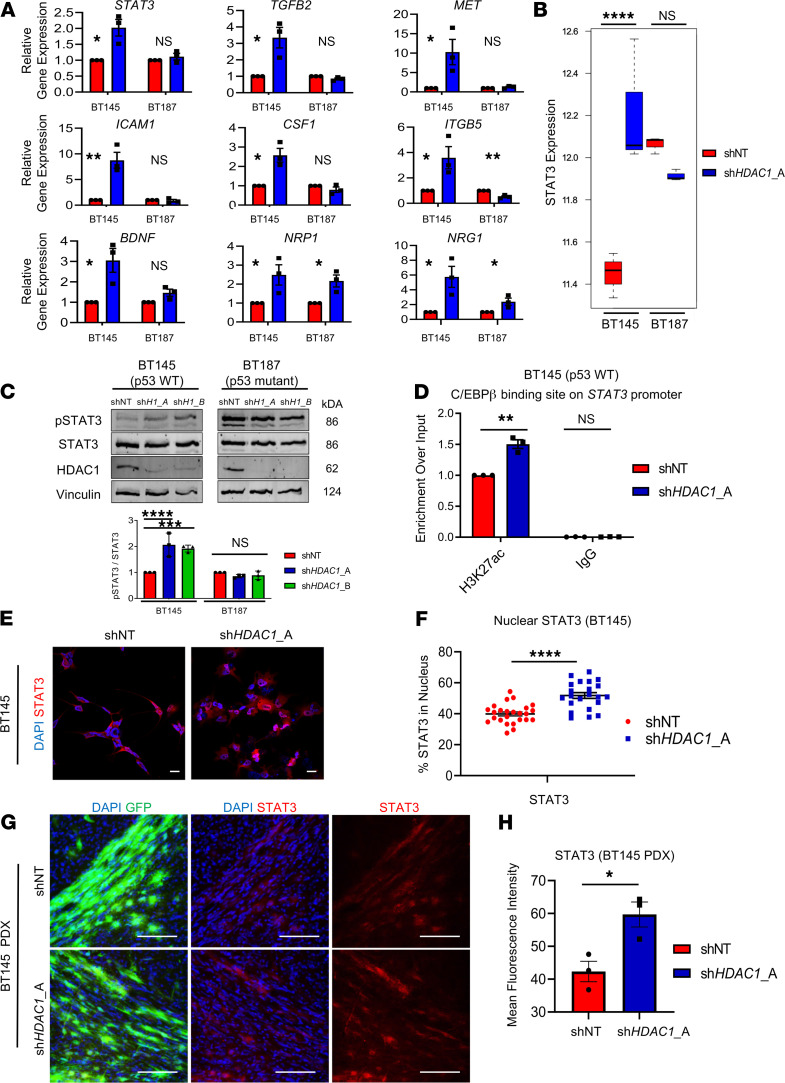
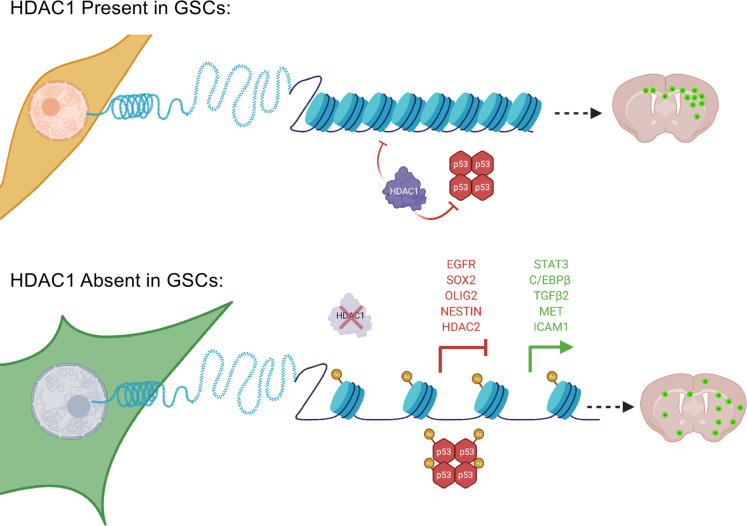
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
