

Somatic PIK3CA Mutations in Sporadic Cerebral Cavernous Malformations
- PMID: 34496175
- PMCID: PMC8606022
- DOI: 10.1056/NEJMoa2100440
Somatic PIK3CA Mutations in Sporadic Cerebral Cavernous Malformations
Abstract
Background: Cerebral cavernous malformations (CCMs) are common sporadic and inherited vascular malformations of the central nervous system. Although familial CCMs are linked to loss-of-function mutations in KRIT1 (CCM1), CCM2, or PDCD10 (CCM3), the genetic cause of sporadic CCMs, representing 80% of cases, remains incompletely understood.
Methods: We developed two mouse models harboring mutations identified in human meningiomas with the use of the prostaglandin D2 synthase (PGDS) promoter. We performed targeted DNA sequencing of surgically resected CCMs from patients and confirmed our findings by droplet digital polymerase-chain-reaction analysis.
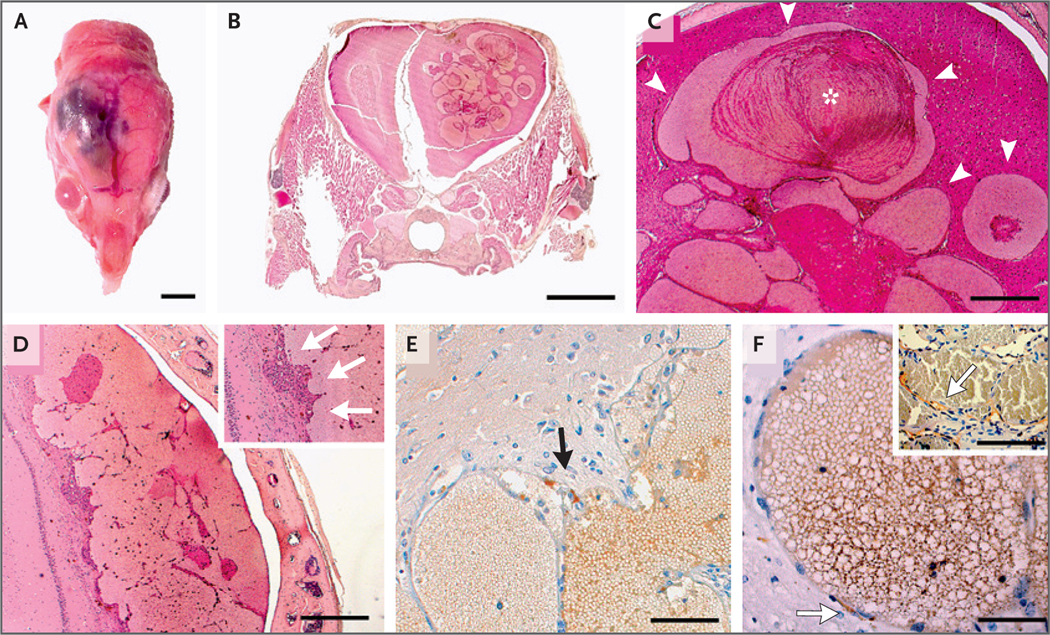
Results: We found that in mice expressing one of two common genetic drivers of meningioma - Pik3ca H1047R or AKT1 E17K - in PGDS-positive cells, a spectrum of typical CCMs develops (in 22% and 11% of the mice, respectively) instead of meningiomas, which prompted us to analyze tissue samples from sporadic CCMs from 88 patients. We detected somatic activating PIK3CA and AKT1 mutations in 39% and 1%, respectively, of lesion tissue from the patients. Only 10% of lesions harbored mutations in the CCM genes. We analyzed lesions induced by the activating mutations Pik3ca H1074R and AKT1 E17K in mice and identified the PGDS-expressing pericyte as the probable cell of origin.
Conclusions: In tissue samples from sporadic CCMs, mutations in PIK3CA were represented to a greater extent than mutations in any other gene. The contribution of somatic mutations in the genes that cause familial CCMs was comparatively small. (Funded by the Fondation ARC pour la Recherche contre le Cancer and others.).
Copyright © 2021 Massachusetts Medical Society.
Figures


Comment in
-
Cellular Origin of Sporadic CCMs.N Engl J Med. 2022 Mar 31;386(13):1290-1291. doi: 10.1056/NEJMc2117812. N Engl J Med. 2022. PMID: 35353971 No abstract available.
-
Cellular Origin of Sporadic CCMs.N Engl J Med. 2022 Mar 31;386(13):1291. doi: 10.1056/NEJMc2117812. N Engl J Med. 2022. PMID: 35353972 No abstract available.
References
-
- Rigamonti D, Hadley MN, Drayer BP, et al. Cerebral cavernous malformations: incidence and familial occurrence. N Engl J Med 1988; 319: 343–7. - PubMed
-
- Riant F, Bergametti F, Ayrignac X, Boulday G, Tournier-Lasserve E. Recent insights into cerebral cavernous malformations: the molecular genetics of CCM. FEBS J 2010; 277: 1070–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous