

Antiviral treatment causes a unique mutational signature in cancers of transplantation recipients
- PMID: 34496298
- PMCID: PMC8516432
- DOI: 10.1016/j.stem.2021.07.012
Antiviral treatment causes a unique mutational signature in cancers of transplantation recipients
Abstract
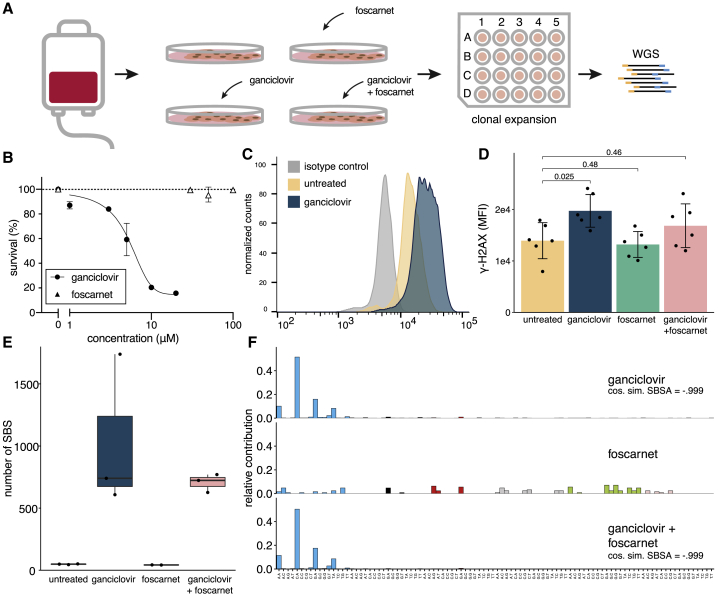
Genetic instability is a major concern for successful application of stem cells in regenerative medicine. However, the mutational consequences of the most applied stem cell therapy in humans, hematopoietic stem cell transplantation (HSCT), remain unknown. Here we characterized the mutation burden of hematopoietic stem and progenitor cells (HSPCs) of human HSCT recipients and their donors using whole-genome sequencing. We demonstrate that the majority of transplanted HSPCs did not display altered mutation accumulation. However, in some HSCT recipients, we identified multiple HSPCs with an increased mutation burden after transplantation. This increase could be attributed to a unique mutational signature caused by the antiviral drug ganciclovir. Using a machine learning approach, we detected this signature in cancer genomes of individuals who received HSCT or solid organ transplantation earlier in life. Antiviral treatment with nucleoside analogs can cause enhanced mutagenicity in transplant recipients, which may ultimately contribute to therapy-related carcinogenesis.
Keywords: antiviral treatment; cancer genomics; cytomegalovirus; ganciclovir; hematopoietic stem cell transplantation; mutational signatures; somatic mutations; therapy-related neoplasms.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests A.R.H., A.v.L., and R.v.B. are named as inventors on a patent application filed resulting from this work.
Figures






Comment in
-
Caught in the antiviral crossfire: Ganciclovir-associated mutagenesis in HSC transplant recipients.Cell Stem Cell. 2021 Oct 7;28(10):1683-1685. doi: 10.1016/j.stem.2021.09.011. Cell Stem Cell. 2021. PMID: 34624229
References
-
- Abascal F., Harvey L.M.R., Mitchell E., Lawson A.R.J., Lensing S.V., Ellis P., Russell A.J.C., Alcantara R.E., Baez-Ortega A., Wang Y. Somatic mutation landscapes at single-molecule resolution. Nature. 2021;593:405–410. - PubMed
-
- Aiuti A., Slavin S., Aker M., Ficara F., Deola S., Mortellaro A., Morecki S., Andolfi G., Tabucchi A., Carlucci F. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. - PubMed
-
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.-L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
